TITLE:High-quality genome assembly and pan-genome studies facilitate genetic discovery in mungbean and its improvement
译名:高质量的基因组组装和泛基因组研究促进了绿豆的基因发现及其改进
期刊:Plant Communications
日期:2022年6月
下载链接: https://doi.org/10.1016/j.xplc.2022.100352

研究介绍:
绿豆(Vigna radiata(L.)R. Wilczek)作为重要的豇豆属成员,具有抗旱、耐贫瘠和固氮能力,同时也是碳水化合物和优质蛋白质以及叶酸和铁等微量元素的重要来源。相对于大豆、菜豆等豆科作物而言,绿豆的基因组研究比较落后,这严重限制了绿豆分子育种的进展。虽然前人研究已经利用二代测序技术组装了一个绿豆基因组草图(VC1973A),并使用GBS等简化基因组测序策略鉴定了一些潜在功能位点。但对于绿豆基因组特征解析、高密度遗传标记、群体多样性差异以及重要功能基因挖掘等研究仍然不足,本研究较为系统地弥补了这些缺陷和空白。
材料与方法:
材料:
本研究从中国绿豆种质资源库的589份种质中筛选出217份具有代表性的种质资源,根据其表型特征、SSR标记的聚类分析结果以及它们的地理分布特征进行分析。将这些材料种植在河北SJZ(114.48E,38.03N)和ZJK(114.88E,40.82N),进行表型观察。总共使用了来自23个国家的750份绿豆种质进行了多样性分析。
方法:
基因组测序和组装
选择了绿豆优良品种JL7进行全基因组测序和组装。提取基因组DNA,在Illumina NovaSeq平台上构建文库并测序。使用KMC(v3.0)估计K-mer分布,用GenomeScope(v2.0)估计基因组大小。对于vRad_JL7基因组的从头组装,使用了基于PacBio Sequel II平台的长读测序。用高分子量(HMW)DNA构建了插入大小约为20kb的DNA文库,并在Novogene有限公司的PacBio Sequel II测序平台上进行了测序。Flye(v2.8.3-b1695)用于从头组装,BWA-MEM(v0.7.17)用于将Illumina PE读数映射到组装序列。在Illumina HiSeq平台上完成HI-C测序文库的构建和测序。使用3D-DNA管道获得染色体规模的组装。最后,使用Juicebox组装工具(v1.11.9)手动更正错误并可视化组装结果。
为了进行基因组评估,使用BWA-MEM将Illumina PE读取映射到vRad_JL7,并使用SamTools(v1.13)和MosDepth(v0.3.1)计算基因组上的短读取覆盖率。然后,使用Busco(v3.1.0)对2326个双子叶植物的单拷贝直系同源基因进行预验证评估。最后,由Ltr_Retriever计算和评估LAI。
基因注释
使用 RepeatModeler和RepeatMasker(v4.0.9)对重复序列进行从头算预测构建了一个TE库,以搜索重复序列注释。BRAKER2管道集成了RNA-seq和基于蛋白质同源性的方法,用于预测蛋白质编码基因。首先,使用 HISAT2(v2.10.2)将 RNA-seq clean reads映射到基因组以获得转录组映射数据。其次,来自 OrthoDB(v10.0)的所有蛋白质序列都被下载并通过 ProtHint(v2.6.0)映射到基因组组装中。然后使用GeneMark-EP+(v4.65)来整合这两种类型的数据。最终的基因结构由Augustus(v3.4.0)预测,非翻译区(UTR)由GUSHR (v1.0) 预测。使用eudicots_odb10的BUSCO来评估注释的质量。我们还使用 Barrnap (v0.9) 预测 rRNA,使用Infernal通过搜索Rfam(v14)预测tRNA和其他非编码RNA数据库。功能注释是通过与公共数据库的同源性搜索分配的,包括SwissProt、NR和基因进化谱系:非监督直系同源组(EggNOG)数据库(E值≤1e-6)。通过从Egg NOG传输注释数据来分配GO和 KEGG注释。Pfam和InterPro基序注释是使用InterProScan(v5.36)分配的。
比较基因组分析
下载了来自以下12个真双子叶植物基因组的蛋白质序列;使用 OrthoFinder(v2.37)鉴定了直系同源基因。使用r8s(v1.81)构建了一个进化树,并进一步使用TimeTree来校准。根据 CAFE(v4.2)的进化树结果与分歧时间和基因家族聚类,通过出生死亡率模型估计每个分支祖先的基因家族成员数,以预测物种rel基因家族的收缩和扩张与他们的祖先有关(P值< 0.05)。GO和KEGG富集分析是使用clusterProfiler(v3.14.0)进行的。随后使用MCScan进行基因组共线性分析,并使用jcvi包中的dotplot脚本可视化同线块。WGDdetector(v1.00)用于通过同义突变率(Ks)方法识别全基因组重复(WGD)。
泛基因组构建与分析
首先,使用MaSuRCA分别组装了217个种质的基因组。然后,通过使用 QUAST,将每个组装的contig与Vrad_JL7对齐,并将长度超过500 nt且同一性小于阈值的contig提取为非参考序列。在比较所有contig的长度和身份之间的关系后,将身份阈值设置为90%。其次,将非参考序列与NT数据库进行比对,去除与微生物、动物和其他非Fabales植物具有良好同源性的序列(E值<1e-5)。然后使用CD-HIT(v4.8.1)对获得的非参考序列进行聚类,以使用90%的同一性阈值去除冗余。使用BLAST将非冗余重叠群与参考基因组进一步对齐。最后,将非冗余和非参考重叠群与参考绿豆基因组合并为绿豆泛基因组。
我们使用“map-to-pan”策略来识别核心和可变基因。使用BWA-MEM和Mosdepth (v0.3.1)将217个绿豆种质与参考基因组进行比对,以估计CDS覆盖率。在整个CDS区域的至少20% 上具有2倍覆盖率的基因被认为存在;否则,他们被视为缺席。通过估计PAV在所有217份材料中的频率,将基因分为两类:“核心”(缺失率 < 0.05)和“可变”基因(缺失率 ≥ 0.05)。 为了估计泛基因组和核心基因组的大小,随机挑选样本(n = 1-217),该过程重复100次。Wilcoxon检验(P值<0.05)用于表示不同亚组基因数差异的显着性水平。
重测序和变异调用
提取基因组DNA并在Illumina NovaSeq平台上对217个绿豆种质进行进一步测序。使用带有默认参数的BWA-MEM将clean reads映射到Vrad_JL7。使用基因组分析工具包 (GATK)(v3.8.1)检测遗传变异,包括SNP和短插入缺失(< 15 bp)。GATK 最终从750 个加入中合并了总共5,671个SNP。根据Vrad_JL7组装的基因模型,使用SnpEff(v5.0e)对上述鉴定的遗传变异进行注释,并且通过VCFtools(v0.1.17)使用500 kb确定沿每个染色体的密度滑动窗口。
种群遗传多样性和结构分析
基于SNP数据,使用ADMIXTURE(v1.3)确定种群结构。 PCA是使用 EIGENSOFT (v6.1.4) 中的 smartpca 函数使用默认参数执行的,并绘制了前两个特征向量。使用R包构建邻接 (NJ)树,并使用 iTOL对树进行可视化。使用 R. Weir中的hclust函数进行基因PAV的种群结构分析,并使用Cockerham的FST估计量来测量多个亚群之间的遗传分化。使用 VCFtools计算FST和全基因组多样性(θπ)的值。
表型和GWAS
大田种植的绿豆种质共观察到33个农艺性状,即19个数量性状和14个离散性状。其中,15个性状(主要是数量性状)在所有6个领域都有记录,另有15个性状只记录一次。对于收集> 3次的性状,平均值用于下游分析。基于SNP和基因PAV数据的GWAS是通过在基因组预证明广泛高效混合模型关联算法(GEMMA) (v0.98.1) 中应用混合线性模型 (MLM) 模型来实现的。来自PCA的前三个主要成分和基因组关系矩阵用于校正种群结构和随机多基因效应。通过过滤P值<0.05并应用调整后的Bonferroni检验来识别重要信号。然后使用的标准评估那些通过阈值的STA和GPTA的稳定性、一致性和稳健性。
结果:
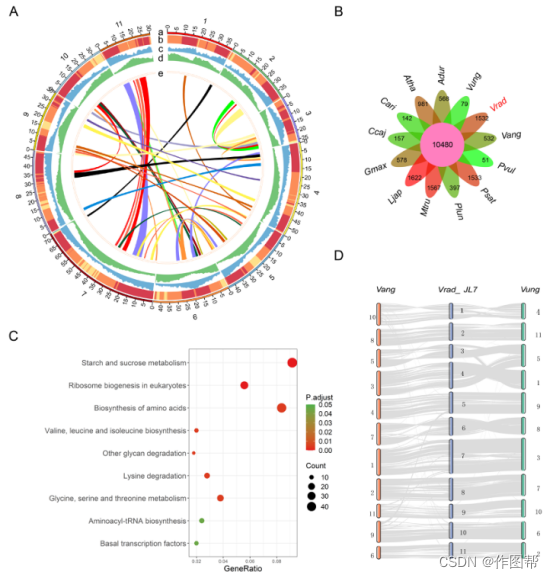
图1、绿豆基因组组装和豆科成员的比较基因组分析。
(A) 绿豆基因组圈图。 从最外到最内,a 假染色体,b GC含量,c 基因分布,d DNA转座子和反转录转座子密度,e 基因组内共线性。
(B) OrthoFinder 鉴定的 12 种豆科和拟南芥的直系同源基因家族。数字代表为每个物种确定的基因家族。
(C) 绿豆中特定基因家族的KEGG通路富集。
(D) Vigna radiata (Vrad_JL7)、Vigna angularis (Vang) 和 Vigna unguiculata (Vung) 之间的基因组共线性。
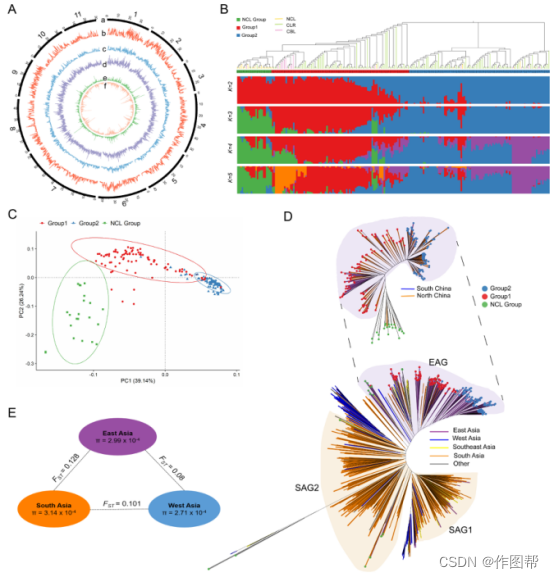
图 2. 绿豆种群基因组分析。
(A)217种材料的变异图谱。圆圈从最外到最内分别代表。a 假染色体;b-d SNP、Indel和SV密度;和e-f非同义和同义SNP。
(B)从核心SNP推断的207个种质的发育树和群体结构(K=2-5)图。绿色、红色和蓝色条带分别代表NCL组、第1组和第2组,树的橙色、绿色和紫色分支分别代表NCL、CLR和CBL。
(C)207个种质的前两个特征向量的PCA图。
(D)207和750个种质的发育树。 树尖的红色、蓝色和绿色分别代表第1组、第2组和NCL组。分支的颜色代表不同的地区。
(E)亚洲不同亚群的F统计量(FST)和核苷酸多样性(π)。
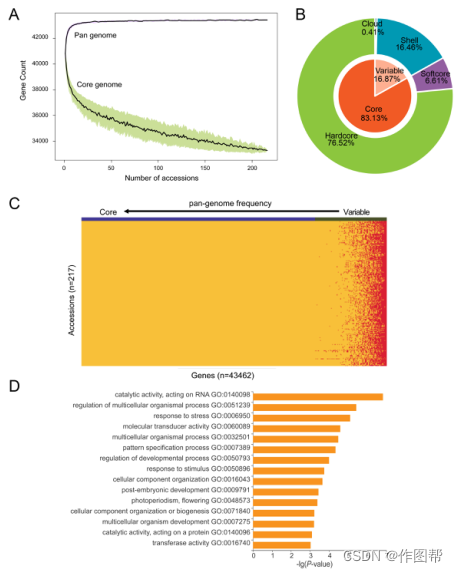
图 3 中国绿豆种质资源泛基因组。
(A) 基于每个给定数量的100个随机组合,模拟泛基因组大小的增加和核心基因组大小的减少。 使用来自所有随机子样本的数据点拟合panand核心基因组曲线,并用黑色实线表示。 紫色和绿色区域的上边缘和下边缘分别对应于最大和最小基因数。
(B) 绿豆泛基因组的组成。
(C) 基因PAV的景观。 基因按其出现频率排序,左侧出现频率最高,右侧出现频率最低。
(D) 可变基因的GO term富集。
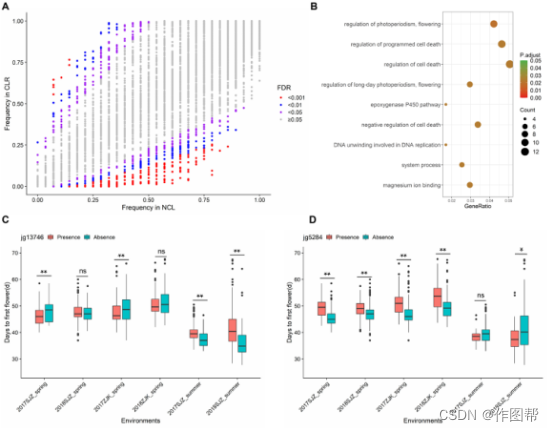
图 4 绿豆从NCLs到CLRs适应过程中选择的基因PAV。
(A) 散点图显示NCL和CLR中的基因出现频率。
(B) 对在适应过程中发生显着表达变化的基因进行KEGG通路富集。
(C和D)2个基因(jg13746和jg5284)在不同环境中调节开花期的存在/不存在变异的例子。 ns、*和**分别表示P值>=0.05、<0.05和<0.01的统计学显着性水平。
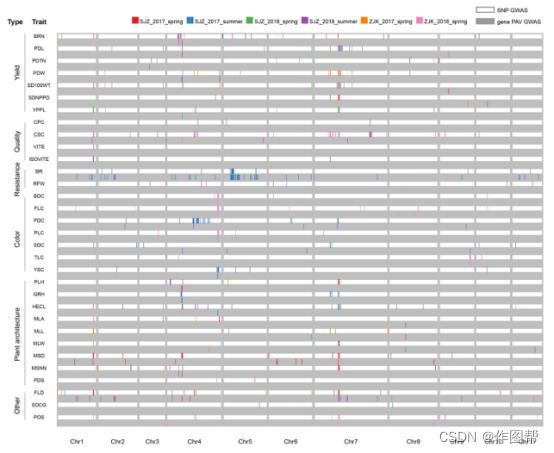
图 5. SNP GWAS和基因PAV GWAS结果总结。
由Vrad_JL7基因组内的SNP GWAS(白框)和基因PAV GWAS(灰框)识别的所有STA和GPTA事件的分布,用于六种不同环境下六种类型的32种性状。
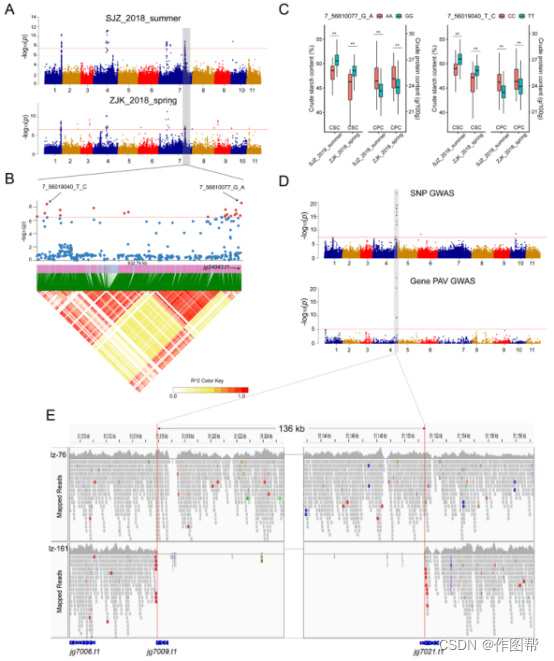
图 6. 粗淀粉含量(CSC)和颜色相关性状的GWAS。
(A)两种不同环境(SJZ_2018_summer和ZJK_2018_spring)中CSC性状的GWAS曼哈顿图。
(B)LD热图显示由 SNP GWAS 识别的强峰周围区域(Chr. 7: 55.979-56.812 Mb)。
(C)两个STA(7_56810077_G_A和7_56019040_T_C)的等位基因与两种环境中的CSC和CPC之间的关系。** 表示 P <0.001的统计显着性水平。
(D)基于SNP的GWAS和基于基因 PAV的GWAS的曼哈顿图,用于颜色相关性状,包括 BDC、FLC、PLC和YSC。
(E)Chr上136 kb 的存在/不存在变体。根据 Integrative Genomics Viewer (IGV)。 样品lz-76(品种:Jilv7)含有此段,颜色为紫色。样品lz-161(品种:VC973A)缺少此段,颜色为绿色或黄色。
结论:
本研究首先利用Pacbio三代测序结合二代测序数据和Hi-C数据进行基因组从头组装,组装出目前为止质量最高的绿豆参考基因组Vrad_JL7,通过对13个近缘物种的基因家族聚类发现,绿豆有1532个特异的基因家族,它们主要与淀粉和糖类代谢、氨基酸合成相关,这与绿豆中丰富的淀粉和蛋白含量是一致的。较基因组显示,绿豆与豇豆属的小豆和豇豆具有很高的共线性,但豇豆的5号染色体分别比对到了绿豆和小豆的两条不同染色体上,说明绿豆、小豆在与豇豆分化后,可能经历了染色体重排事件。群体结构和PCA分析都能将材料明显地划分为三组,且SNP与gene PAV的结果高度一致。研究进一步调查了217份材料的33个重要农艺性状在6个环境的表型,部分性状之间具有很高的相关性,基于SNP的GWAS分析显示,多个性状的STAs聚集在1、4、7号染色体上的热点区域,它们可能与绿豆的驯化选择有关。发掘出jg24043等一系列候选位点,并在大豆等近缘物种中能找到对应的同源基因。这些候选基因为绿豆分子育种改良提供了重要基础。