数据质控、质检是所有生信分析必不可少的步骤,GWAs分析的质控大抵可分为7个步骤:控制检出率(Missing)、剔除性别错误(Sex Discrepancy)、控制次等位基因频率(MAF)、去除反哈达——温伯格平衡(HWE)项、控制杂合率(Heterozygosity Rates)、控制样本关系(血缘关系)、控制群体分层(Population Stratification)。
一、背景简介
本内容参考AndriesT. Marees等方法(DOI:10.1002/mpr.1608),使用的程序包为PLINK v1.9,二进制数据来自 International HapMap Project的模拟数据(祖先来自欧洲西北部的犹他州居民),包含三个二进制数据:“.bed”,包含所有患者和健康对照的基因型信息(次文件内容为二进制数据,方便计算机读取,不便于肉眼查看)。“.fam”,包含研究个体的谱系关系(父、母本)、性别和表型信息等。“.bim”,包含SNPs的位置信息(Table 1)。
Table 1:PLINK支持的二进制文件
| 后缀 | 内容信息 |
|---|---|
| .bed | 实验所有个体及对照的基因型信息 |
| .fam | 个体及对照的谱系关系、性别和表型信息 |
| .bim | SNPs的位置信息 |
除二进制文件外,PLINK还支持文本文件(Table 2)。但是,相对于二进制文件,PLINK处理文本文件速度较慢,故更推荐使用二进制文件。
Table 2:PLINK支持的文本文件
| 后缀 | 内容信息 |
|---|---|
| .ped | 基因型信息、个体信息 |
| .map | 遗传标记信息 |
两种数据格式的数据排布形式如下(Fig. 1)。
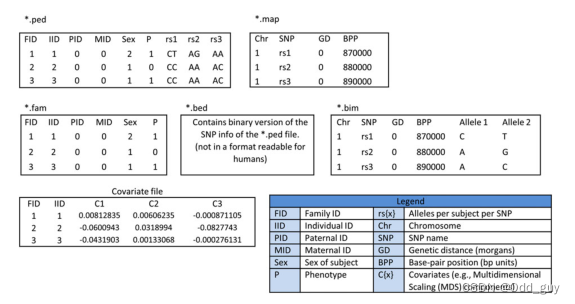
Fig. 1:PLINK数据排布形式
二、详细步骤
在开始之前需要从PLINK官网下载PLINK v1.9并解压。(记住解压目录)
#创建GWAs目录,用于存放所有相关文件
mkdir GWAs
#下载PLINK
wget https://s3.amazonaws.com/plink1-assets/plink_linux_x86_64_20220402.zip
#解压PLINK包
unzip plink_linux_x86_64_20220402.zip
下载PLINK后需获取模拟数据集。可直接从AndriesT. Marees的Github主页克隆此项目的工作目录(Fig. 2)。并将目录下的压缩包解压。
#克隆项目
git clone https://github.com/MareesAT/GWA_tutorial.git
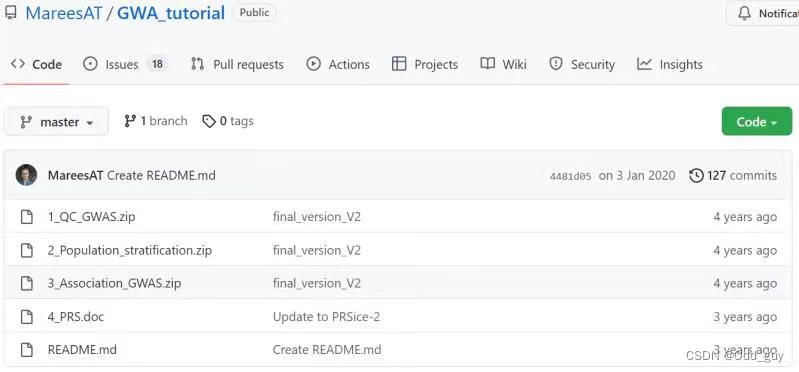
Fig. 2:GWA_tutorial项目
1、控制缺失率(检出率)
(1)创建工作目录
PLINK每次运作都会生成多个文件,所以我喜欢把每一步的文件放到一个独立的目录(GWAs)中。
#将GWA_tutorial目录下的文件全部移动到GWAs目录下
cd GWA_tutorial
mv * /{your directory}/GWAs/
#解压三个压缩包
unzip 1_QC_GWAS.zip
unzip 2_Population_stratification.zip
unzip 3_Association_GWAS.zip#删除GWA_tutorial目录
cd ..
rm -r GWA_tutorial#创建存放控制检出率文件的目录
cd /{your directory}/GWAs/1_QC_GWAS/
mkdir missing
#将所需文件放入missing目录
mv HapMap_3_r3_1.* missing/
mv hist_miss.R missing/
(2)质检
同时检查数据集中的数据缺失(SNPs和个体)。
/{your directory}/GWAs/plink-20220402/plink --bfile HapMap_3_r3_1 --missing
| 输出文件 | 内容信息 |
|---|---|
| plink.lmiss | 缺失个体信息的SNPs(后续绘图需要) |
| plink.imiss | 缺失SNPs的个体(后续绘图需要) |
| plink.hh | 杂合单倍体基因型列表(所有file.hh文件) |
| plink.log | 本次运行的工作日志 |
(3)绘图
使用R绘制检出率直方图(根据plink.lmiss和plink.imiss)。输出为两个PDF文件,可查看检出率(SNPs或个体)的频率分布。
#绘制直方图
Rscript --no-save hist_miss.R
(4)质控
从数据集中移除数据缺失率(missingness)过高项。过滤的阈值可以在一开始设置较大,第二步时再设置较小的阈值,便于设置合适的阈值。
PS:必须先过滤缺失个体信息SNPs,再过滤缺失SNPs的个体。
#删除missingness > 0.2的SNPs
/{your directory}/GWAs/plink-20220402/plink --bfile HapMap_3_r3_1 --geno 0.2 --make-bed --out HapMap_3_r3_2
#删除missingness > 0.2的个体
/{your directory}/GWAs/plink-20220402/plink --bfile HapMap_3_r3_2 --mind 0.2 --make-bed --out HapMap_3_r3_3#设置一个更小的阈值(0.02),进一步过滤数据
/{your directory}/GWAs/plink-20220402/plink --bfile HapMap_3_r3_3 --geno 0.02 --make-bed --out HapMap_3_r3_4
/{your directory}/GWAs/plink-20220402/plink --bfile HapMap_3_r3_4 --mind 0.02 --make-bed --out HapMap_3_r3_5
(5)最终数据集
HapMap_3_r3_5.bed
HapMap_3_r3_5.bim
HapMap_3_r3_5.fam
2、去除性别错误
(1)创建工作目录
创建工作目录(sex_dis),存放过滤性别错误(Sex Discrepancy)的文件。
#回到数据过滤的主工作目录
cd /{your directory}/GWAs/1_QC_GWAS/
#创建工作目录(sex_dis)并移入所需文件
mkdir sex_dis
cp missing/HapMap_3_r3_5.* sex_dis/
mv gender_check.R sex_dis/
(2)质检
在PLINK中,基于X染色体同源性评估(Homozygosity Estimate)的F值确定每个个体的生物学性别,对于数据集记录的性别和生物学性别不一致的个体会被标记为“PROBLEM”项,在后续的质控中被移除或修正。
检察并标记PROBLEM项。
cd sex_dis
#检查并标记PROBLEM
/{your directory}/GWAs/plink --bfile HapMap_3_r3_5 --check-sex
通过工作日志文件plink.log可知发现有一个PROBLEM项(Fig. 3),可打开输出文件plink.sexcheck查看。
vi plink.log
vi plink.sexcheck
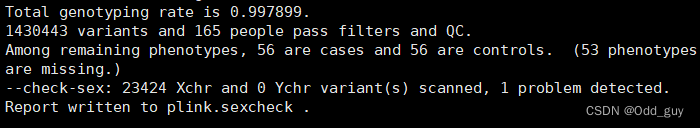
Fig. 3:plink.log
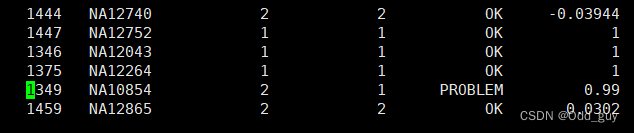
Fig. 4:plink.sexcheck
(3)绘图
调用R可视化检查结果(Fig. 5),在女性个体中有一个错误项。
Rscript --no-save gender_check.R
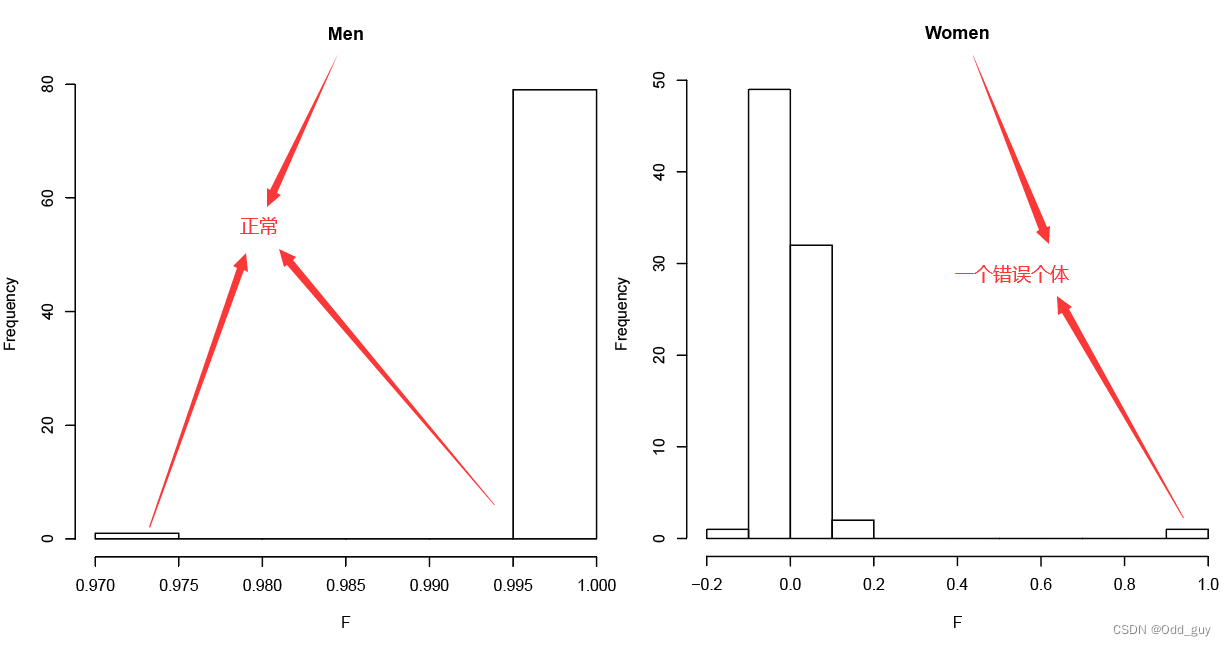
Fig. 5:性别检查结果
(4)质控
抽取性别错误项。
#生成包含PROBLEM标记的行前两列信息的列表
grep "PROBLEM" plink.sexcheck| awk '{print$1,$2}'> sex_discrepancy.txt
脚本解析:grep可按关键字遍历列表,匹配带有“PROBLEM”字符串的列,awk可处理文本文件,此处将匹配得到的行的前两列信息写入文件sex_discrepancy.txt。
常见的方法有两种,一种是直接移除错误项,另一种是将错误项的性别按遗传(生物学)性别更改。个人更推荐直接移除性别错误项,因为修改数据可能伴随有不必要的麻烦发生。
方法1:根据sex_discrepancy.txt从数据集中移除错误项。
#根据sex_discrepancy.txt从数据集中移除错误项
/{your dierctory}/GWAs/plink-20220402/plink --bfile HapMap_3_r3_5 --remove sex_discrepancy.txt --make-bed --out HapMap_3_r3_6
方法2:将错误项的性别按遗传(生物学)性别更改。
#将错误项的性别按遗传(生物学)性别更改
/{your dierctory}/GWAs/plink-20220402/plink --bfile HapMap_3_r3_5 --impute-sex --make-bed --out HapMap_3_r3_6
(5)最终数据集
HapMap_3_r3_6.bed
HapMap_3_r3_6.bim
HapMap_3_r3_6.fam
3、控制次等位基因频率
(1)创建工作目录
创建工作目录(MAF)存放此步骤相关文件。
cd /{your directory}/GWAs/1_QC_GWAS/
#创建工作目录
mkdir MAF
#将相关文件存放到MAF目录下
cp sex_dis/HapMap_3_r3_6.* MAF/
mv MAF_check.R MAF/
(2)质检
在开始此步骤之前需要将数据集中除常染色体外的SNPs移除。
#进入工作目录
cd MAF
#匹配1~22号(常)染色体,并将对应的SNPs编号写入snp_1_22.txt
awk '{ if ($1 >= 1 && $1 <= 22) print $2 }' HapMap_3_r3_6.bim > snp_1_22.txt
#基于snp_1_22.txt从数据集中提取常染色体SNPs
/{your directory}/GWAs/plink-20220402/plink --bfile HapMap_3_r3_6 --extract snp_1_22.txt --make-bed --out HapMap_3_r3_7
检查数据集中的次等位基因频率(Minor Allele Frequency,MAF),并调用R绘图。
#质检
/{your directory}/GWAs/plink-20220402/plink --bfile HapMap_3_r3_7 --freq --out MAF_check
(3)绘图
#基于MAF_check.frq绘图
Rscript --no-save MAF_check.R
查看pdf文件(Fig. 6),根据样本的大小和MAF的总体分布情况确定过滤阈值,常用的阈值为0.1或0.05。此处设置阈值为0.05。
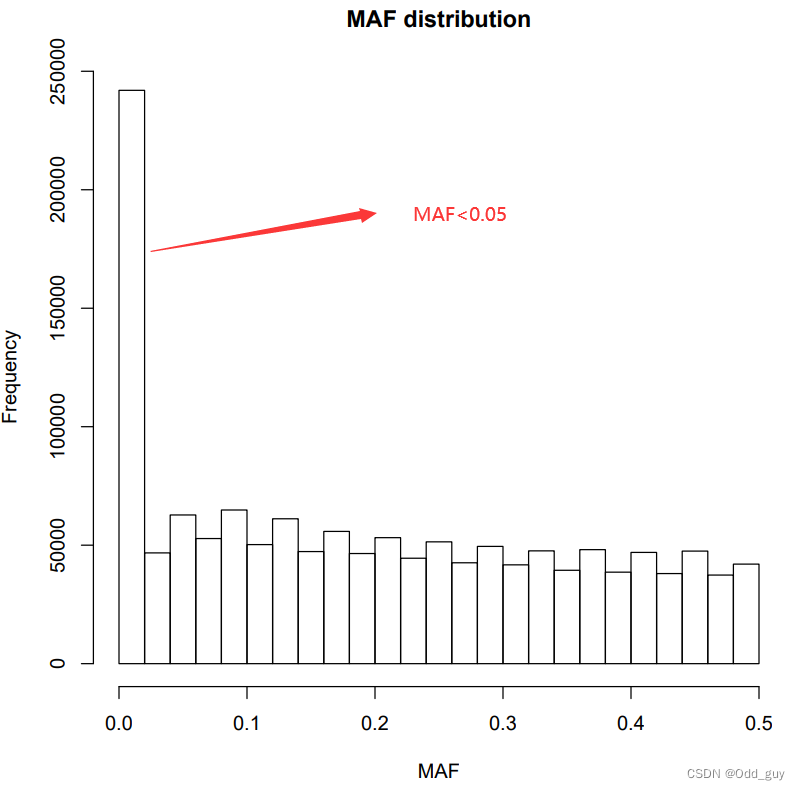
Fig. 6:MAF分布
(4)质控
删除MAF < 0.05的SNPs。
#质控
/{your directory}/GWAs/plink-20220402/plink --bfile HapMap_3_r3_7 --maf 0.05 --make-bed --out HapMap_3_r3_8
查看此次运行的工作日志可知,有325,318个位点被过滤;有164个个体,包含1,073,226个位点保留。
(5)最终数据集:
HapMap_3_r3_8.bed
HapMap_3_r3_8.bim
HapMap_3_r3_8.fam
4、去除反哈达——温伯格平衡项
(1)创建工作目录
创建工作目录HWE,置入所需文件。
cd /{your directory}/GWAs/1_QC_GWAS/
mkdir HWE
cp MAF/HapMap_3_r3_8.* HWE/
mv hwe.R HWE/
(2)质检
计算哈达——温伯格(HWE)对应的P值。
#计算HWE的P值
/{your directory}/GWAs/plink-20220402/plink --bfile HapMap_3_r3_8 --hardy
(3)绘图
从上一步生成的表格plink.hwe中提取P < 0.00001的项,将其写入plinkzoomhwe.hwe。分别基于两个文件绘图(Fig. 7)。
#提取提取**P < 0.00001**的项
awk '{ if ($9 <0.00001) print $0 }' plink.hwe>plinkzoomhwe.hwe
#绘图
Rscript --no-save hwe.R
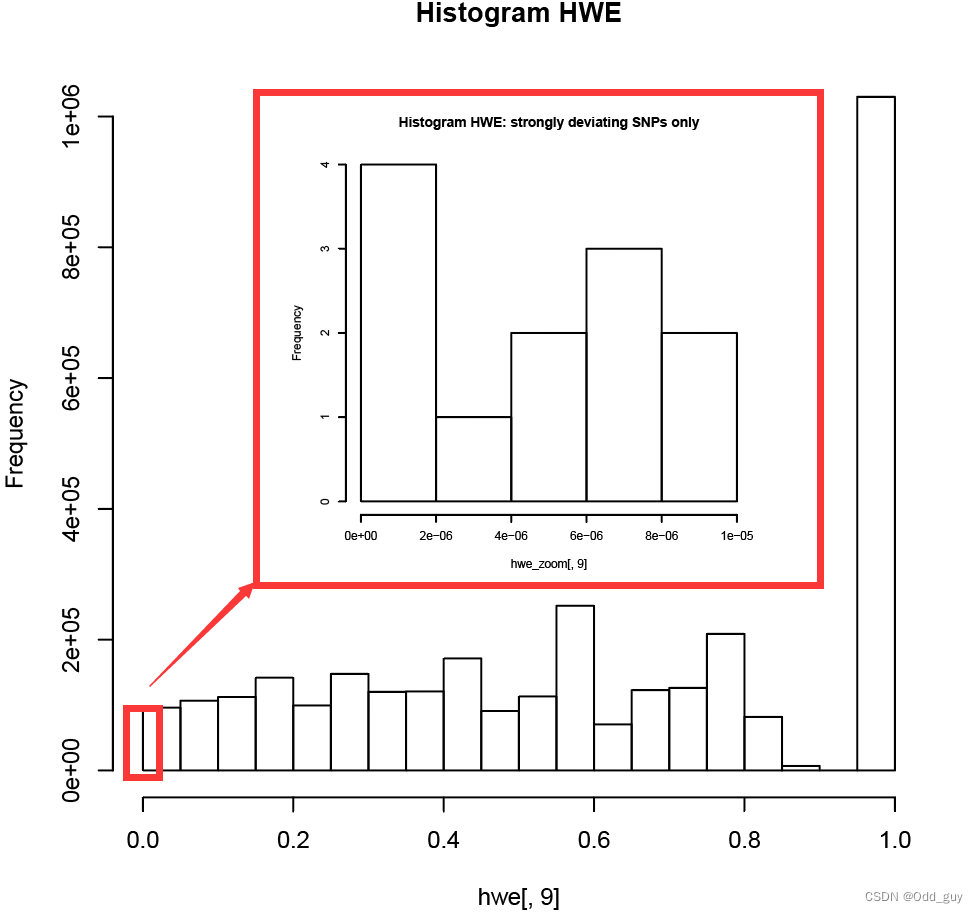
Fig. 7:HWE P值分布
(4)质控
为了避免过滤掉与性状相关的数据,可以先使用一个较大的(较严格)阈值(P < 1e-6)过滤对照样本,再设置一个较小的(较宽松)的阈值(P < 1e-10)来过滤所有样本。
#过滤对照组
/{your directory}/GWAs/plink-20220402/plink --bfile HapMap_3_r3_8 --hwe 1e-6 --make-bed --out HapMap_hwe_filter_step1#过滤所有样本
/{your directory}/GWAs/plink-20220402/plink --bfile HapMap_hwe_filter_step1 --hwe 1e-10 --hwe-all --make-bed --out HapMap_3_r3_9
(5)最终数据集
HapMap_3_r3_9.bed
HapMap_3_r3_9.bim
HapMap_3_r3_9.fam
5、控制杂合率
(1)创建工作目录
创建工作目录。
#创建工作目录Het_rate
cd /{your directory}/GWAs/1_QC_GWAS/
mkdir Het_rate
#置入文件
cp HWE/HapMap_3_r3_9.* Het_rate
mv check_heterozygosity_rate.R Het_rate
mv inversion.txt Het_rate
mv heterozygosity_outliers_list.R Het_rate
(2)质检——基于连锁不平衡裁剪数据集
首先,我们需要在排除了高反转区域(文件inversion.txt提供)的前提下,检出相关系数并不高的SNPs,并将它们写入列表indepSNP.prune.in(此文件后续需要用到)。
#进入工作目录
cd Het_rate
#检出相关系数不高的SNPs
/{your directory}/GWAs/plink-20220402/plink --bfile HapMap_3_r3_9 --exclude inversion.txt --range --indep-pairwise 50 5 0.2 --out indepSNP
但是在PLINK 1.9中,形参“–range”已经被移除,可以将指令修改为以下形式(Fig. 8)。
#针对PLINK 1.9,检出相关系数不高的SNPs
/{your directory}/GWAs/plink-20220402/plink --bfile HapMap_3_r3_9 --exclude range inversion.txt --indep-pairwise 50 5 0.2 --out indepSNP
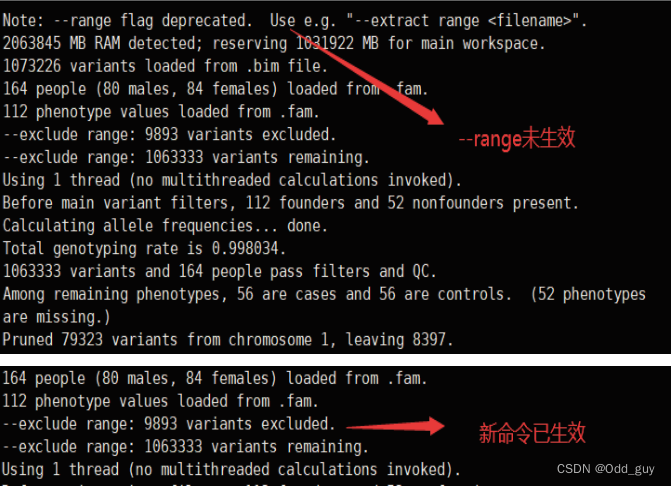
Fig. 8:参数更改
此步骤中提供给形参“–indep-pairwise”的实参含义为:SNPs窗口大小50,成对计算R2值(LD:连锁不平衡);如果R2大于0.2则删除其中一个SNP,并将窗口向前步移5个位点,以此类推。
关于拟合度:此时的拟合度表示SNP-SNP两两之间的连锁程度(Fig. 9)。

Fig. 9:连锁程度
输出的文件中,indepSNP.prune.out包含被移除的SNPs列表,indepSNP.prune.in包含余下的SNPs列表。记录在log文件中的信息表明,总计有1,063,333个位点,其中959,189个位点被移除。
(3)绘图
提取剩余SNPs所在个体的相关信息。
#将剩余SNPs所在个体及其相关信息写入R_check.het
/{your directory}/GWAs/plink-20220402/plink --bfile HapMap_3_r3_9 --extract indepSNP.prune.in --het --out R_check
R_check.het中包含的信息见下表(Table 3)。
Table 3:关键信息
| 列名 | 包含信息 |
|---|---|
| O(HOM) | 实际纯合子数 |
| E(HOM) | 预测纯合子数 |
| N(NM) | 总基因数 |
| F | 近交系数 |
绘制杂合率频率分布直方图(Fig . 10)。
R脚本中杂合率计算: H a t . r a t e = ( N − O ) N Hat.rate=\frac{(N-O)}{N} Hat.rate=N(N−O)
Rscript --no-save check_heterozygosity_rate.R
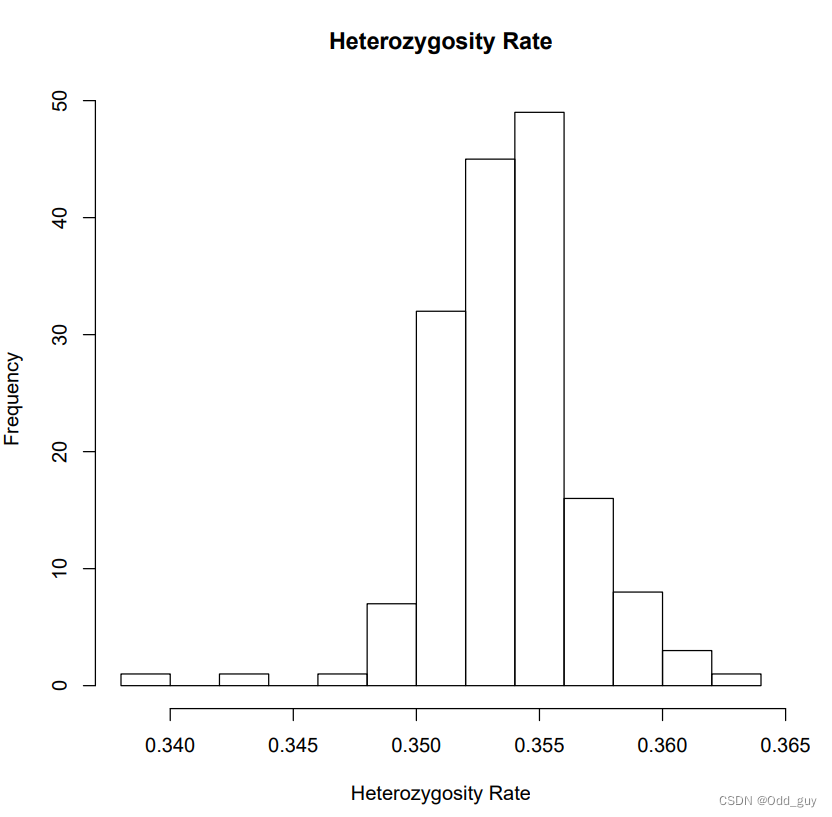
Fig. 10:杂合率直方图
(4)质检——标记离群值
使用R脚本遍历列表,并将离群值列表(使用拉依达法检出)写入fail-het-qc.txt。
#使用R抽取离群值
Rscript --no-save heterozygosity_outliers_list.R
R代码及解析:
#读取文件
het <- read.table("R_check.het", head=TRUE)
#计算杂合率作为HET_RATE列插入列表
het$HET_RATE = (het$"N.NM." - het$"O.HOM.")/het$"N.NM."
#筛选杂合率大于或小于三倍标准差的个体,构成列表het_fail
#统计学中,均值周围三倍标准差范围可包括99.73%的观测值
het_fail = subset(het, (het$HET_RATE < mean(het$HET_RATE)-3*sd(het$HET_RATE)) | (het$HET_RATE > mean(het$HET_RATE)+3*sd(het$HET_RATE)));
#计算杂合距离,作为HET_DST列插入列表het_fail
het_fail$HET_DST = (het_fail$HET_RATE-mean(het$HET_RATE))/sd(het$HET_RATE);
#将列表het_fail写入文件fail-het-qc,txt
write.table(het_fail, "fail-het-qc.txt", row.names=FALSE)
(5)质控
fail-het-qc.txt文件无法直接被PLINK 1.9识别(R在读取字符串时会自动添加双引号),需要对其进行处理。
sed 's/"// g' fail-het-qc.txt | awk '{print$1, $2}'> het_fail_ind.txt
脚本解析:sed是一种流编辑器(Stream Editor),在此处将列表中的双引号全部删除(使用命令’s/"// g’匹配和删除双引号),即使用第二根斜线后的内容替换第二根斜线前的内容。
移除离群个体(两个)。
/{your directory}/GWAs/plink-20220402/plink --bfile HapMap_3_r3_9 --remove het_fail_ind.txt --make-bed --out HapMap_3_r3_10
(6)最终数据集
HapMap_3_r3_10.bed
HapMap_3_r3_10.bim
HapMap_3_r3_10.fam
6、控制样本关系
并不是所有的实验都需要此步骤,或者说可以针对实验目的和样本的谱系结构设计特定的过滤方法。
(1)创建工作目录
创建工作目录
#创建工作目录
cd /{your directory}/GWAs/1_QC_GWAS/
mkdir Relatedness#置入文件
mv Relatedness.R Relatedness
cp Het_rate/HapMap_3_r3_10.* Relatedness/
cp Het_rate/indepSNP.prune.in Relatedness/
(2)质检
对亲缘关系的检查是基于血缘同源(Identity By Descent,IBD)和pihet(Proportion IBD),IBD为两个个体有相同且来自同一祖先alleles,pihet和IBD的关系为: p i h a t = P ( I B D = 2 ) + pihat={P_{(IBD=2)}}+ pihat=P(IBD=2)+ 1 2 1\over2 21 P ( I B D = 1 ) {P_{(IBD=1)}} P(IBD=1),用于表征两个个体的亲缘关系,取值0~1,pihat越大关系越近;而状态同源(Identity By States,IBS)表示两个个体有相同的alleles。
检出个体间pihat > 0.2的项,即假设在一个随机的样本的中需去除这些项:
#计算IBD,检出pihat > 0.2的项
/{your directory}/GWAs/plink-20220402/plink --bfile HapMap_3_r3_10 --extract indepSNP.prune.in --genome --min 0.2 --out pihat_min0.2
结果(Fig. 11)以列表的形式被写入pihat_min0.2.genome,关键信息见Table 4:
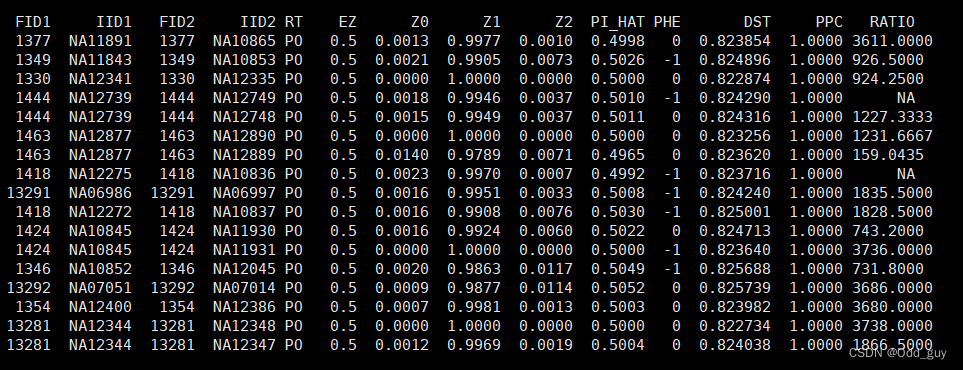
Fig. 11:pihat_min0.2.genome
Table 4:IBD计算结果列名
| 列名 | 含义 |
|---|---|
| RT | 数据集中记录的关系类型 |
| EZ | 基于数据集记录的IBD共享期望值 |
| Z0 | P ( I B D = 0 ) _{(IBD=0)} (IBD=0) |
| Z1 | P ( I B D = 1 ) _{(IBD=1)} (IBD=1) |
| Z2 | P ( I B D = 2 ) _{(IBD=2)} (IBD=2) |
| PI_HAT | Proportion IBD |
| PHE | 表型编码,1对应AA,0表示AU,-1表示UU |
| DST | IBS距离: I B S . d i s = IBS.dis= IBS.dis= I B S 2 + 0.5 I B S 1 N ( S N P p a i r ) IBS2+0.5IBS1\over N_{(SNP pair)} N(SNPpair)IBS2+0.5IBS1 |
| PPC | IBS二项式检验 |
| RATIO | IBS0 SNP率 |
筛选 P ( I B D 1 ) > 0.9 P_{(IBD1)} > 0.9 P(IBD1)>0.9 的项。
#遍历列表,筛选Z1 > 0.9的项,并将其写入zoom_pihat.genome
awk '{ if ($8 >0.9) print $0 }' pihat_min0.2.genome>zoom_pihat.genome
(3)绘图
使用R脚本Relatedness.R绘图。
#绘图
Rscript --no-save Relatedness.R
R脚本解析:
#绘制所有计算的项
pdf("relatedness.pdf")
#读取文件
relatedness = read.table("pihat_min0.2.genome", header=T)
#设置点的大小形状
par(pch=16, cex=1)
#建立坐标系
with(relatedness,plot(Z0,Z1, xlim=c(0,1), ylim=c(0,1), type="n"))
#添加RT列为PO的点
with(subset(relatedness,RT=="PO") , points(Z0,Z1,col=4))
#添加RT列为UN的点
with(subset(relatedness,RT=="UN") , points(Z0,Z1,col=3))
#添加图例
legend(1,1, xjust=1, yjust=1, legend=levels(relatedness$RT), pch=16, col=c(4,3))#绘制Z1 > 0.9的项
pdf("zoom_relatedness.pdf")
#读取文件
relatedness_zoom = read.table("zoom_pihat.genome", header=T)
#设置点的大小和形状
par(pch=16, cex=1)
#设置坐标系,x&y轴依据relatedness.pdf结果调整
with(relatedness_zoom,plot(Z0,Z1, xlim=c(0,0.02), ylim=c(0.98,1), type="n"))
#添加RT列为PO的点
with(subset(relatedness_zoom,RT=="PO") , points(Z0,Z1,col=4))
#添加RT列为UN的点
with(subset(relatedness_zoom,RT=="UN") , points(Z0,Z1,col=3))
#添加图例
legend(0.02,1, xjust=1, yjust=1, legend=levels(relatedness$RT), pch=16, col=c(4,3))#绘制pihat直方图
pdf("hist_relatedness.pdf")
#读取文件
relatedness = read.table("pihat_min0.2.genome", header=T)
#绘图
hist(relatedness[,10],main="Histogram relatedness", xlab= "Pihat")
dev.off()
结果(直方图未列出)见Fig. 12,依据RT列画图,蓝色点(PO)表示有亲代-子代关系的pair,红色点(UN)表示没有关系的pair。关于RT列的取值见Table 5。
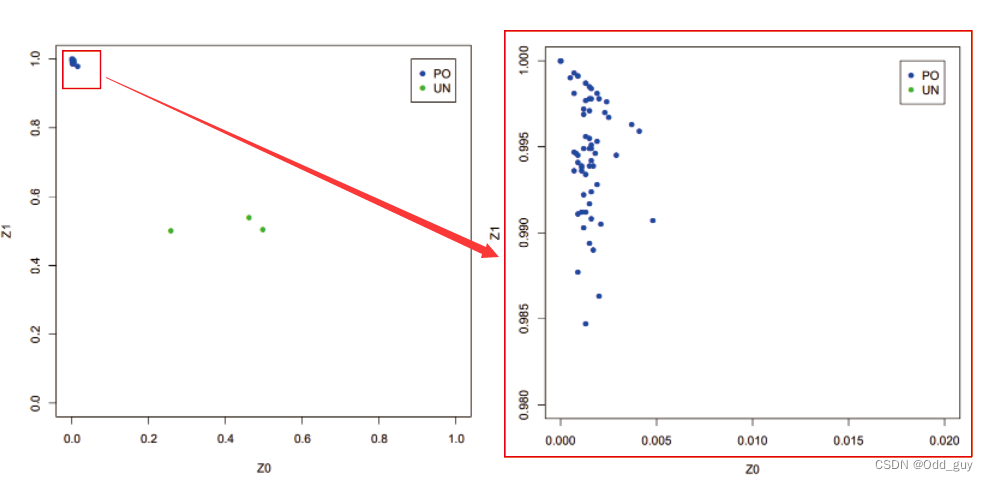
Fig. 12:Relatedness可视化
Table. 5:RT列取值
| 取值 | 含义 |
|---|---|
| OR | 亲代-子代关系 |
| UN | 无关系 |
| FS | 同胞,有相同的亲本 |
| HS | 半同胞,同母异父或同父异母 |
| OT | 其他 |
(4)验证质检结果
为了验证检出的pair都是“亲本-子代”(OR)关系,我们可以提取founders(亲本不在数据集内的个体)。
#提取founders
/{your directory}/GWAs/plink-20220402/plink --bfile HapMap_3_r3_10 --filter-founders --make-bed --out HapMap_3_r3_11
再次对founders检出(pihat > 0.2)。
#仅对founders数据集进行检出
/{your directory}/GWAs/plink-20220402/plink --bfile HapMap_3_r3_11 --extract indepSNP.prune.in --genome --min 0.2 --out pihat_min0.2_in_founders
查看输出结果pihat_min0.2_in_founders.genome(Fig. 13),可发现,在移除含有亲本-子代关系的个体后,仅对founders进行检出,其中pihat大于0.2的pair只有一项,其出现的原因可能是信息记录错误。说明之前检出的pihat大于0.2的个体的确都是由于亲代-子代关系。

Fig. 13:只对founders结果进行检出的结果
(5)质检
文章中提供的办法是移除pair(Fig. 13)中检出率较高的个体(Fig. 14),但是个人觉得为避免误差,需将二者共同移除(因为无法确定是那个个体记录错误),但是为了和文章提供的教程保持一致性,仍按教程进行。
#再次计算检出率
/{your directory}/GWAs/plink-20220402/plink --bfile HapMap_3_r3_11 --missing
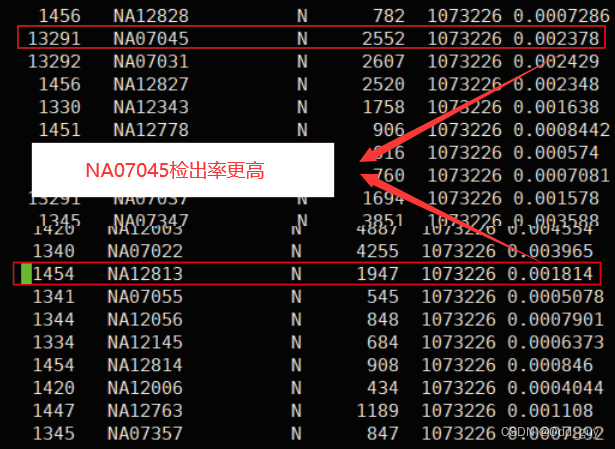
Fig. 14:再次计算检出率结果
为了提供移除检出个体的参考,创建一个名为“0.2_low_call_rate_pihat.txt”的文件,文件内包含列表:
13291 NA07045
参考0.2_low_call_rate_pihat.txt内的列表,对数据集进行质控,即移除关系不是“亲代-子代”关系,且pihat值大于0.2的pair中检出率较高的个体(13291 NA0704)。
#移除质检不合格个体
/{your directory}/GWAs/plink-20220402/plink --bfile HapMap_3_r3_11 --remove 0.2_low_call_rate_pihat.txt --make-bed --out HapMap_3_r3_12
(6)最终数据集
HapMap_3_r3_12.bed
HapMap_3_r3_12.bim
HapMap_3_r3_12.fam
7、控制群体分层(结构)
见后续。
Ending