初探QTL和GWAS
文章目录
- 初探QTL和GWAS
- 实验目的
- 实验内容
- 实验题目
- 第一题:玉米MAGIC群体的QTL分析
- 第二题:TASSEL自带数据集的关联分析
- 实验过程
- 玉米MAGIC群体的QTL分析
- ① 包含的数据
- ② 绘制LOD曲线
- 株高对应的QTL
- TASSEL自带数据集的关联分析
- TASSEL简介
- 实际操作
- 曼哈顿图和QQ图
- TOP5位点
- 讨论
- 1.QTL分析和GWAS分析的一般流程是什么?
- 2.QTL分析中,如何确定目标性状的候选遗传位点?
- 3.GWAS分析中,如何确定目标性状的候选遗传位点?如何缩小候选区间?
- 4.如何降低QTL、GWAS分析结果的假阳性?
- 4.如何降低QTL、GWAS分析结果的假阳性?
实验目的
- 了解R语言工作环境
- 熟悉QTL和GWAS分析的一般流程
- 掌握至少一种常用QTL和GWAS分析软件
实验内容
- 使用R包
qtl2进行QTL分析 - 使用软件
TASSEL进行GWAS分析 - 熟悉基因型、表型等数据格式,掌握QTL和GWAS分析结果的解读和可视化
实验题目
第一题:玉米MAGIC群体的QTL分析
利用玉米MAGIC群体数据,使用R/qtl2完成以下QTL分析:
① 本群体包含哪些数据?
② 绘制株高(PH)的LOD曲线
③ 给出株高对应的QTL
数据来源参考网址:
qtl2data/maize_magic.zip at main · rqtl/qtl2data (github.com)
第二题:TASSEL自带数据集的关联分析
使用TASSEL对自带数据集(安装目录下TutorialData子目录中),对其它两个性状(EarHT和EarDia)中任意一个,进行关联分析。
至少需要给出以下结果:
①与EarHT或EarDia最显著关联的Top 5位点信息(包括染色体号、位置和P值)
②两张图(曼哈顿图、QQ图)及相应的解释(绘图结果说明什么)
实验过程
玉米MAGIC群体的QTL分析
① 包含的数据
maize_magic_geno.csv: MAGIC的基因型数据
Maize_magic_foundergeno.csv: 9个founder系的基因型数据
Maize_magic_gmap.csv: 遗传图谱
Maize_magic_pmap.csv: 物理图谱
Maize_magic_crossinfo.csv: cross信息(代数后面跟着表示九个founder相对贡献的值)
Maize_magic_pheno.csv: 表型数据
maize_magic_phenocovar.csv: 表现型协变量,只描述表现型
② 绘制LOD曲线
我们使用R语言的qtl2包,进行QTL定位分析。qtl2的使用方法可以参考官方文档,写的很不错。
R/qtl2 user guide (kbroman.org)
下面就是代码实现了,注释写的也挺清楚的。
# 设置工作目录
setwd("D:/00大三上/生信原理/实践作业/QTL_GWAS")# install.packages("qtl2")
library(qtl2)
# 直接从网站上读取
file <- paste0("https://raw.githubusercontent.com/rqtl/","qtl2data/main/MaizeMAGIC/maize_magic.zip")MM <- read_cross2(file)
# 或者把压缩包下载下来再读取
MM <- read_cross2("./maize_magic.zip")# 观察数据 - 10条染色体、总计41324个标记
summary(MM)# (伪)标记插入遗传图,获取假定QTL;以1cM为间隔插入伪标记
MMmap <- insert_pseudomarkers(map=MM$gmap, step=1)# 计算基因型概率;假定基因分型误差概率0.002
# cores=4,使用多核计算
MMpr <- calc_genoprob(cross = MM, map = MMmap, error_prob=0.002, cores=4)# 可视化查看基因型概率
# 参数依次为 基因型概率、marker图、要查看的个体编号、要查看的染色体号
# 染色体的坐标在横轴上,基因型在纵轴上。较高的基因型概率表示为暗色
png(filename = 'plot_genoprob.png',width = 3580,height = 2200,res = 300)
plot_genoprob(MMpr, MMmap, ind = 1, chr = 1)
dev.off()# 运用 Haley-Knott regression 进行基因组扫描
# 可加协变量、此处不加。输出LOD分数矩阵(positions × phenotypes)
MM_scan1_out <- scan1(MMpr, MM$pheno, cores=0)# 绘制LOD曲线,指定一列(表型)
png(filename = 'plot_scan1.png',width = 3580,height = 2200,res = 300)
plot_scan1(MM_scan1_out, map = MMmap, lodcolumn = "PH")
dev.off()
# 查看对PH而言哪个伪标记LOD分数最高、哪个基因型标记LOD得分最高
sort(MM_scan1_out[,"PH"])# permutation test 说明scan结果的统计学意义
# 识别随机下可能出现的最大LOD分数,使用1000种排列
# 这一行执行时间比较长,因为置换检验需要重新计算1000次
MM_operm <- scan1perm(genoprobs = MMpr, pheno = MM$pheno, n_perm = 1000) # 显著性阈值、默认5%水平
# PH为7.40 期望LOD得分低于7.40是偶然事件
summary(MM_operm, alpha=0.05) # 寻峰,95%置信区间
thr <- summary(MM_operm)
MM_peaks <- find_peaks(scan1_output = MM_scan1_out, map = MMmap, threshold = thr, prob = 0.95, expand2markers = FALSE)# 查看PH表型对应多少满足阈值的峰(QTL),分别在哪
PH_qtl <- MM_peaks[MM_peaks$lodcolumn=='PH',]# 保存结果,方便之后查看
write.csv(thr,"thr.CSV")
write.csv(PH_qtl,"PH_qtl.CSV")
![[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-A75Zs1cH-1671348220695)(D:/typora%E5%9B%BE%E7%89%87/plot_genoprob.png)]](https://img-blog.csdnimg.cn/115b45372f79463eb3a1eaca78c5f4da.png)
![[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-UxcOenMR-1671348220696)(D:/typora%E5%9B%BE%E7%89%87/plot_scan1.png)]](https://img-blog.csdnimg.cn/4f84085e6a6840c895cf0fecd06ecfff.png)
由LOD曲线可以看出,6、8和10号染色体很有可能有QTL位点,这也与下文的分析对应上了。
株高对应的QTL
有上面代码的输出结果(表1、表2)可以看到,PH的阈值是7.41,筛选过后可以得到三个QTL。分别位于6、8、10号染色体上,这也与上面的LOD曲线对应上了。
| PS | PH | EH | GYrad | |
|---|---|---|---|---|
| 0.05 | 7.424502 | 7.408967 | 7.753835 | 7.628907 |
| lodcolumn | chr | pos | lod | ci_lo | ci_hi | |
|---|---|---|---|---|---|---|
| 4 | PH | 6 | 12.28468 | 15.91047 | 11.0098 | 12.3023 |
| 5 | PH | 8 | 90.14981 | 10.9792 | 79.44594 | 93.54998 |
| 6 | PH | 10 | 13.94991 | 7.916526 | 9.585541 | 43.14987 |
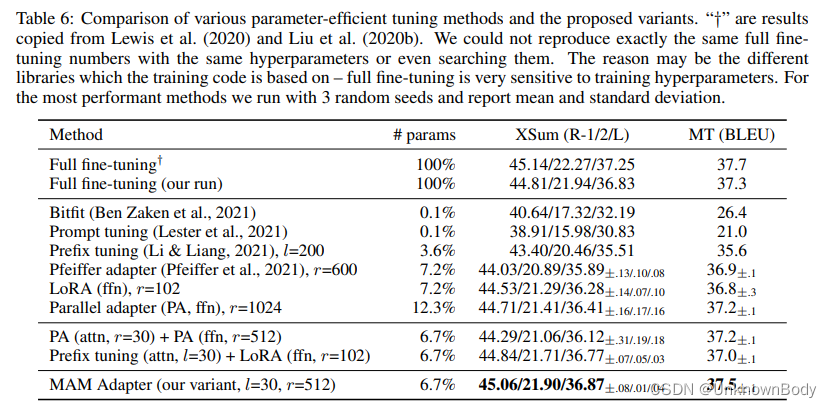
TASSEL自带数据集的关联分析
TASSEL简介
Trait Analysis by aSSociation, Evolution and Linkage.
TASSEL是一个软件包,用于评估特征的关联,进化模式和连锁不平衡。本软件的优点包括:
-
有机会使用一些新的强大的统计方法来进行关联映射,例如通用线性模型(GLM)和混合线性模型(MLM)。MLM是《Nature Genetics》论文—— Unified Mixed-Model Method for Association Mapping,该技术减少了与复杂谱系、家族、创始效应和种群结构关联映射中的I型错误。
-
能够处理广泛的索引(插入和删除)。大多数软件忽略了这种类型的多态性;然而,在某些物种(如玉米)中,这是最常见的多态性类型。
Maize Genetics | TASSEL
实际操作
下载安装完TASSEL后,打开安装目录下TASSEL5\TutorialData中的示例数据。
分别导入:
- 基因型数据mdp_genotype.hmp
- 群体结构数据 mdp_population_strucutre
- 表型性状数据 mdp_phenotype、mdp_traits
![[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-LhqZsYwn-1671348220697)(D:/typora%E5%9B%BE%E7%89%87/image-20221217185130743.png)]](https://img-blog.csdnimg.cn/3972fe62cd8b485fa031279fe57df7d0.png)
菜单栏 的Filter可以基于每个位点的基因型统计分析,进行筛选。
我们通过MAF来进行筛选。
Minor Allele Frequency (MAF):次等位基因频率
比如:某个SNP位点,包含A和G两种等位基因(allele),100个个体中有35个为A,65个为G,则该位点的MAF=0.35
过滤完成后,软件界面左侧会出现文件mdp_genotype_Filter
![[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-jag7bR1G-1671348220698)(D:/typora%E5%9B%BE%E7%89%87/image-20221217185311242.png)]](https://img-blog.csdnimg.cn/8a34cbde5a654f479d863dd839b9e875.png)
![[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-k9ZRC19S-1671348220698)(D:/typora%E5%9B%BE%E7%89%87/image-20221217185818287.png)]](https://img-blog.csdnimg.cn/55e5b3461a014b74b24f48c4d343f1e8.png)
数据过滤后,我们计算群体内不同个体之间的亲缘关系,可用于后续混合线性模型MLM的关联分析。
![[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-visY6s9a-1671348220699)(D:/typora%E5%9B%BE%E7%89%87/image-20221217190829676.png)]](https://img-blog.csdnimg.cn/015024bb0f5f4eebaea6cf9f8e826a4e.png)
选择默认的Kinship计算方法,Center IBS和Max Alleles使用默认值。
![[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-OCJk31YV-1671348220699)(D:/typora%E5%9B%BE%E7%89%87/image-20221217190939874.png)]](https://img-blog.csdnimg.cn/fe6be3b3ffb141c3aba4b93e0d103caa.png)
下表是有关参数设置的帮助。
| Parameter | Description | Values | Default |
|---|---|---|---|
| Kinship method | The Centered_IBS (Endelman - previously Scaled_IBS) method produces a kinship matrix that is scaled to give a reasonable estimate of additive genetic variance. Uses algorithm http://www.g3journal.org/content/2/11/1405.full.pdf Equation-13. The Normalized_IBS (Previously GCTA) uses the algorithm published here: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3014363/pdf/main.pdf. | Centered _IBS, Normalized _IBS, Dominance _Centered _IBS, Dominance _Normalized _IBS | Centered _IBS |
| Max Alleles | Max Alleles | 2…6 | 6 |
| Algorithm Variation | Algorithm Variation | Observed _Allele _Freq, Proportion _Heterozygous | Observed _Allele _Freq |
计算完成后,生成亲缘关系矩阵。
![[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-a3v1XDqK-1671348220700)(D:/typora%E5%9B%BE%E7%89%87/image-20221217191158236.png)]](https://img-blog.csdnimg.cn/0c15e23dee754261955972b93fb2dfac.png)
![[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-wfdTXN0N-1671348220700)(D:/typora%E5%9B%BE%E7%89%87/image-20221217201532859.png)]](https://img-blog.csdnimg.cn/205d37aea7594d2da34c4275352fb966.png)
![[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-rjRUQWTo-1671348220700)(D:/typora%E5%9B%BE%E7%89%87/image-20221217195848466.png)]](https://img-blog.csdnimg.cn/b34cf2b7f4544478a472e720f3676a76.png)
去掉最后一个群体结构(Q3),指导手册给出的解释说,如果我们把它们全部作为协变量使用,这会产生线性相依性。
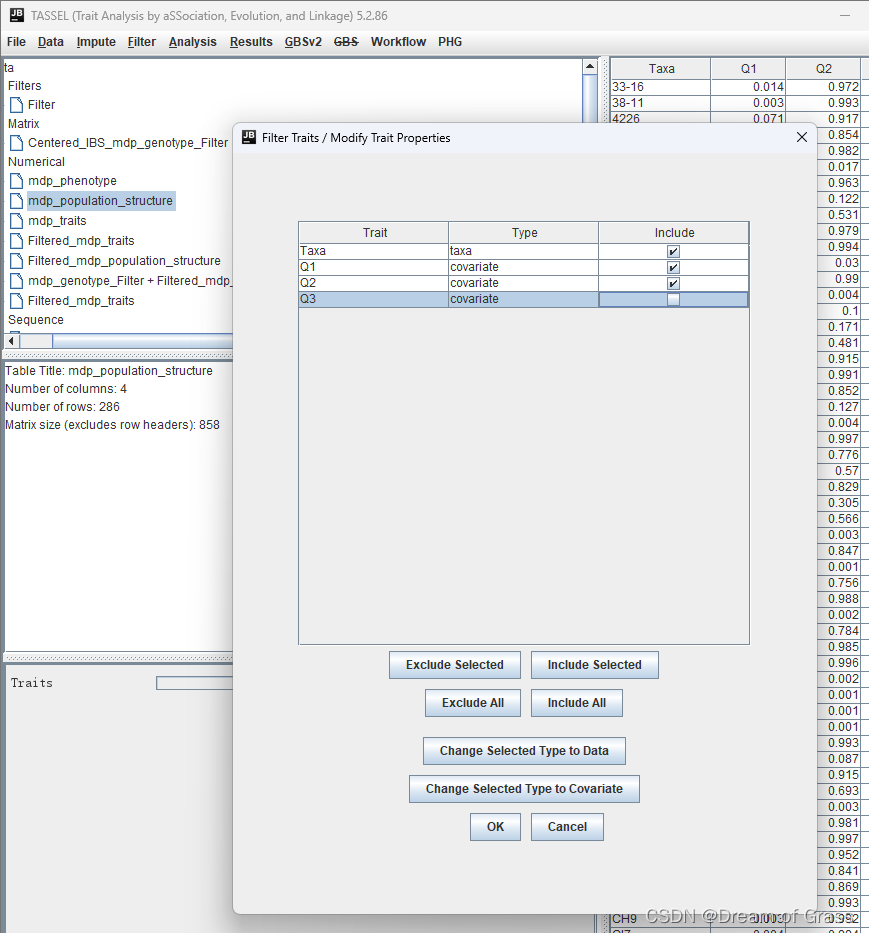
摁住Ctrl键同时选中上述三个文件( mdp_genotype_Filter,Filtered_mdp_traits ,Filtered_mdp_population_structure )进行合并, 点击Data → Intersect Join
![[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-JrBDGCpg-1671348220701)(D:/typora%E5%9B%BE%E7%89%87/image-20221217200052155.png)]](https://img-blog.csdnimg.cn/884d8e39ffb24f15a5a3321baba316cc.png)
![[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-Z6cKtN21-1671348220701)(D:/typora%E5%9B%BE%E7%89%87/image-20221217200112457.png)]](https://img-blog.csdnimg.cn/fd67986fcc694d6289be051089758ae2.png)
使用GLM进行关联分析(默认参数)。
![[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-d97fBYJ8-1671348220702)(D:/typora%E5%9B%BE%E7%89%87/image-20221217200145457.png)]](https://img-blog.csdnimg.cn/7cd5983e68ab45c9bf9311ed7a8ce8e1.png)
![[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-IM76vlh6-1671348220702)(D:/typora%E5%9B%BE%E7%89%87/image-20221217200203731.png)]](https://img-blog.csdnimg.cn/7779368d83e44852a94fa99b583a0ca4.png)
曼哈顿图和QQ图
得到结果之后,我们就可以产看结果了,在菜单栏里的Results里面有画曼哈顿图和QQ图的选项。
曼哈顿图中每个点代表一个SNP,纵轴为每个SNP计算出来的Pvalue取-log10,横轴为SNP所在的染色体。基因位点的Pvalue越小即-log10(Pvalue)越大,其与表型性状或疾病等关联程度越强。
可以发现1、8和10号染色体与EarTH的性状可能有关联,从QQ图上也可以看到,偏离了对角线,有一个凸起,表明有些标记可能是与EarTH性状相关。
![[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-Kj5PvckS-1671348220703)(D:/typora%E5%9B%BE%E7%89%87/glm_manhattan-1671290211488-9.png)]](https://img-blog.csdnimg.cn/fe6eb8dbc08741b9aee782b147c8bbe5.png)
![[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-ksjxyAmz-1671348220703)(D:/typora%E5%9B%BE%E7%89%87/glm_qq-1671290490486-11.png)]](https://img-blog.csdnimg.cn/5825b9718b6f484b8a7a6c007e9c21f9.png)
接下来我们再试试混合线性模型,操作和GLM差不多。
![[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-i8ieCsNW-1671348220704)(D:/typora%E5%9B%BE%E7%89%87/image-20221217201745695.png)]](https://img-blog.csdnimg.cn/5d1ff0cbc1434cb493f4f97af512efed.png)
从QQ图来看,和性状关联的位点不多,故后面分析用GLM模型的结果。
![[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-oGK3v0ap-1671348220704)(D:/typora%E5%9B%BE%E7%89%87/mlm_manhattan-1671290839598-13.png)]](https://img-blog.csdnimg.cn/14af0a9b1d2145f381a50f73912a4a2f.png)
![[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-D1cgWd0W-1671348220705)(D:/typora%E5%9B%BE%E7%89%87/mlm_qq-1671290845234-15.png)]](https://img-blog.csdnimg.cn/1cfd87a0101c42d9924892e8ed97edc8.png)
TOP5位点
我们将p值从小到大排序则可以看到最显著关联的5个位点。
![[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-FocrHSql-1671348220705)(D:/typora%E5%9B%BE%E7%89%87/image-20221217202223015.png)]](https://img-blog.csdnimg.cn/4c94e92804574fc6a1dcd5d64823bbfb.png)
导出之后的表格如下
| Chr | Pos | p |
|---|---|---|
| 10 | 33619582 | 0.000016724 |
| 1 | 245136244 | 0.000021302 |
| 4 | 194749287 | 0.000028892 |
| 4 | 194749230 | 0.000048693 |
| 8 | 134813437 | 0.000052242 |
讨论
1.QTL分析和GWAS分析的一般流程是什么?
QTL分析的一般流程包括以下步骤:
- 构建分离群体(作图群体)
- 获取每个个体的目标性状的表型数据
- 对作图群体进行基因分型:获取个体基因型数据、筛选分子标记
- 使用统计方法检测QTL
QTL分析的一般流程包括以下步骤:
- 自然群体资源收集和鉴定
- 获取目标性状的表型数据
- 群体重测序(全基因组、转录组、外显子组、GBS等),获取基因型数据(测序读段比对、遗传变异如SNP的识别、GWAS变异标记(如SNP)构建)
- 基因型补缺和过滤,表型数据分析
- 遗传多样性、群体结构、亲缘关系、LD等分析
- 关联分析
- 候选基因筛选与挖掘
2.QTL分析中,如何确定目标性状的候选遗传位点?
在QTL分析中,目标性状的候选遗传位点通常使用统计方法来识别,该方法分析个体样本中遗传变异和性状之间的关系。
有几种方法可用于识别候选qtl,包括:
- 连锁分析:该方法利用家族中遗传标记(如snp)的遗传模式来识别可能与性状相关的位点。
- 关联分析:这种方法在不相关的个体样本中测试遗传变异和性状之间的统计关联。
- 混合模型分析:该方法结合了连锁和关联分析的元素,以解释可能影响性状的已知和未知因素的影响。
一旦确定了候选qtl,就可以使用其他数据和方法进一步验证它们,例如在不同的样本中重复研究或使用功能测定来确定关联背后的生物学机制。考虑潜在的混杂因素也很重要,比如环境或生活方式因素,这些因素可能会影响这一特征。
3.GWAS分析中,如何确定目标性状的候选遗传位点?如何缩小候选区间?
在GWAS分析中,通常使用统计方法分析遗传变异与样本中个体的目标特征之间的关系,从而确定目标特征的候选遗传位点()。
可以使用以下几种方法来确定候选GWAS位点:
- 单标记分析:对于每个单个的位点(例如SNP)进行统计分析,以识别与特征关联的位点。
- 多标记分析:对于一组位点进行统计分析,以识别与特征关联的位点。
- 基于统计模型的分析:使用统计模型来考虑可能影响特征的各种因素(包括环境和生活方式因素)。
确定候选GWAS位点后,可以使用附加数据和方法进一步验证它们,例如在不同样本中复制研究或使用功能性分析来确定关联的生物学机制。
要缩小候选区间,可以使用多种方法,包括:
- 增加样本大小:使用更多的个体来提高统计力,使候选区间更加明确。
- 使用高分辨率基因分型技术:使用高分辨率基因分型技术(如下一代测序)可以提供更多的遗传变异数据,从而帮助缩小候选区间。
- 引入附加信息:使用其他数据(如生物学或功能性分析结果)来帮助缩小候选区间。
- 复制研究:在不同的样本中复制研究可以帮助确定候选区间的稳定性。
- 使用多种统计方法:使用多种不同的统计方法可以帮助缩小候选区间,因为不同的方法可能会得出不同的结论。
4.如何降低QTL、GWAS分析结果的假阳性?
选择重组事件多、变异丰富、群体分化不大的群体。并且选择合适的模型。
候选GWAS位点后,可以使用附加数据和方法进一步验证它们,例如在不同样本中复制研究或使用功能性分析来确定关联的生物学机制。
要缩小候选区间,可以使用多种方法,包括:
- 增加样本大小:使用更多的个体来提高统计力,使候选区间更加明确。
- 使用高分辨率基因分型技术:使用高分辨率基因分型技术(如下一代测序)可以提供更多的遗传变异数据,从而帮助缩小候选区间。
- 引入附加信息:使用其他数据(如生物学或功能性分析结果)来帮助缩小候选区间。
- 复制研究:在不同的样本中复制研究可以帮助确定候选区间的稳定性。
- 使用多种统计方法:使用多种不同的统计方法可以帮助缩小候选区间,因为不同的方法可能会得出不同的结论。
4.如何降低QTL、GWAS分析结果的假阳性?
选择重组事件多、变异丰富、群体分化不大的群体。并且选择合适的模型。