大家好,这里是专注表观组学十余年,领跑多组学科研服务的易基因。
全基因组重亚硫酸盐测序(WGBS)是甲基化研究的重要技术。尽管已经开发了一系列工具来解决由亚硫酸盐处理引起的比对问题,但尚未对最新可用工具的reads比对性能以及多种哺乳动物的生物学解释(biological interpretation)进行评估。在此基础上,本文对人、牛和猪等三种哺乳动物真实和模拟WGBS生成的14.77 billion reads数据进行936次比对,以测试和评估14种常用的标准分析比对软件,分别为Bwa-meth、BSBolt、BSMAP、Walt、Abismal、Batmeth2、Hisat_3n、Hisat_3n_repeat、Bismark-bwt2-e2e、Bismark-his2、BSSeeker2-bwt、BSSeeker2-soap2、BSSeeker2-bwt2-e2e和BSSeeker2-bwt2-local。
具体而言,与其他九种比对软件相比,Bwa-meth、BSBolt、BSMAP、Bismark-bwt2-e2e和Walt五种软件表现出更高的唯一比对reads、比对精确率、召回率和F1-score,不同比对软件对甲基化的影响在CpG位点数量和甲基化水平、calling差异甲基化CpG(DMC)和差异甲基化区域(DMR)上有很大差异。尤其是BSMAP比对软件在检测CpG位点和甲基化水平、DMC/DMR/DMR相关基因和信号通路的calling方面表现出最高的准确性。

图1:哺乳动物14种比对软件的标准分析方案
材料方法
(1)模拟数据的生成
从UCSC下载了人、牛和猪的参考基因组,其基因组大小、组装质量和基因数量各不相同(表1)。基于三种哺乳动物的参考基因组,使用Sherman生成模拟的WGBS数据(Sherman是Babraham开发的具有特定参数的WGBS数据模拟器)。
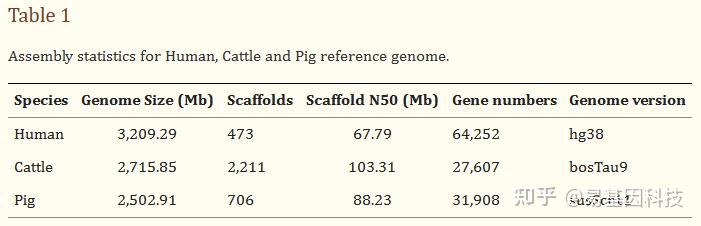
表1:人、牛和猪参考基因组组装
模拟对运行时间、占用内存、唯一比对reads、比对精确率、召回率和F1-score进行标准分析测试,使用Sherman模拟数据集A生成了45个模拟数据样本(5个测序错误率×3个哺乳动物×3个重复)。5种测序错误率分别为0、0.25%、0.5%、0.75%和1.00%;3种哺乳动物为人、牛和猪;每种哺乳动物的每个测序错误率生成3个重复。总共使用了45个模拟数据样本和630次比对来对14个比对软件进行标准测试。每个模拟数据样本均生成2M reads覆盖,因此45个模拟数据样本共生成90M reads。
为进一步研究比对性能以及重复序列和CGI对比对软件比对率的影响,又生成了包含18个数据样本的模拟数据集B(2个测序错误率×3个哺乳动物×3个重复),每个样本的reads数约为93.8M。2个测序错误率分别为0和1.00%,共生成约1.64 billion reads数据。
两组模拟数据集研究均未对包括CpG位点、DMC/DMR/DMR相关基因和信号通路calling的甲基化生物学解释进行标准分析。
(2)真实WGBS数据
从国家生物技术信息中心(NCBI)数据库的序列reads档案中下载了人、牛和猪的真实WGBS数据。真实数据集A包含9个数据样本(3个哺乳动物×3个重复)。每个样本覆盖2M reads,共生成18M reads,用于评估运行时间、内存消耗和唯一比对reads。
真实数据集B包含18个数据样本(3个哺乳动物×2组×3个重复)。两组分为第1组和第2组,每组3个生物学重复。人的数据来自精神分裂症患者和正常人的大脑;牛的数据来自6头公牛背最长肌;猪的数据来自长白猪的骨骼肌。真实数据集B中样本的测序深度大于30X,共生成13.1 billion reads用于分析比对软件对生物学解释下游分析的影响,如CpG位点、DMC/DMR/DMR基因和信号通路。
分析结果
(1)比对软件的运行时间和内存使用成本
为了对这14种比对软件的比对效率进行标准测试分析,使用模拟数据集A和真实数据集A计算运行时间和内存消耗。结果发现Walt软件速度最快,而BSSeeker2-bwt2-local是在测序错误率分别为0%、0.25%、0.50%、0.75%和1.00%下的最慢软件(图2a)。Bwa-meth、Hisat_3n、Hisat_3n_repeat、BSBolt、BSMAP和Bismark-his2的运行时间与人、牛和猪这三种哺乳动物中分别为0、0.25%、0.50%、0.75%和1.00%的测序错误率呈正相关(Pearson相关系数≥0.7,P<0.05)(图2b)。BSSeeker2-bwt2-e2e、Batmeth2、BSSeeker2-bwt2-local和Abismal的平均运行时间比Walt、BSMAP、Bwa-meth、Bismark-his2、BSSeeker2-soap2、BSSeeker2-bwt、Bismark-bwt2-e2e、BSBolt、Hisat_3n和Hisat_3n_repeat的平均运行时间分别长5.38倍(模拟数据)和3.36倍(真实数据)(学生t检验,P<2.67e-12)(图2c)。

图2:基于模拟数据集A和真实数据集A的14种比对软件的运行时间和内存消耗。
模拟数据集A中五种测序错误率的14种比对软件的运行时间(左)和内存消耗(右)。
模拟数据集A中14种比对软件的测序错误率与性能之间的Pearson相关系数(如运行时间、内存消耗、唯一比对reads,比对的精确率和召回率)。
模拟数据集A(左)和真实数据集A(右)中14种比对软件的平均运行时间。
模拟数据集A(左)和真实数据集A(右)中14种比对软件的平均内存消耗。
将模拟数据和实际数据结果进行平均,除了Batmeth2、BSSeeker2-bwt2-local和Abismal软件外,其他比对软件的运行时间在人、牛和猪物种之间没有显著变化(图2c)。BSSeeker2-soap2软件分别比BSSeeker2-bwt、BSSeeker2-bwt2-e2e、BSSeeker2-bwt2-local软件快2.72、5.78和8.95倍(学生t检验,P<3.7e-09);Bismark-his2软件比Bismark-bwt2-e2e软件快2.27倍(学生t检验,P<9.5e-07);Bismark-bwt2-e2e软件比BSSeeker2-bwt2-e2e软件快2.04倍(学生t检验,P<2.3e-09),表明不同的比对软件表现出不同的运行时间。
此外,在分别为0、0.25%、0.50%、0.75%和1.00%的测序错误率中,Walt占用内存最多,而BSSeeker2-bwt2-local占用内存最低,这与Walt和BSSeeker2-bwt2-local的运行时间不一致(图2a)。Bwa-meth和BSBolt所占内存与人、牛和猪分别为0、0.25%、0.50%、0.75%和1.00%的测序错误率高度正相关(Pearson相关系数≥0.7,P<0.05,图2b)。BSSeeker2-soap2、BSMAP、Bwa-meth、Walt、Batmeth2、Hisat_3n、Hisat_3n_repeat和BSBolt软件在人类、牛和猪中所占内存分别是BSSeeker2-bwt、Bismark-bwt2-e2e、BSSeeker2-bwt2-e2e、BSSeeker2-bw2-local、Bismark-his2和Abismal的3.29倍(模拟数据)和3.30倍(真实数据)(学生t检验,P<2.2e-16,图2d)。且这些比对软件的内存随着人、牛和猪的基因组大小而减少(图2d)。将模拟数据和真实数据的结果进行平均,BSSeeker2-soap2所占内存分别是BSSeeker2-bwt、BSSeeker2-bwt2-e2e和BSSeeker2-bwt2-local的2.19倍、2.02倍和2.02倍;Bismark-his2所占内存是Bismark-bwt2-e2e的1.33倍(学生t检验,P<7.37e-06)。这些结果表明,soap2和his2比对软件比bwt、bwt2-e2e和bwt2-local占用更多内存。
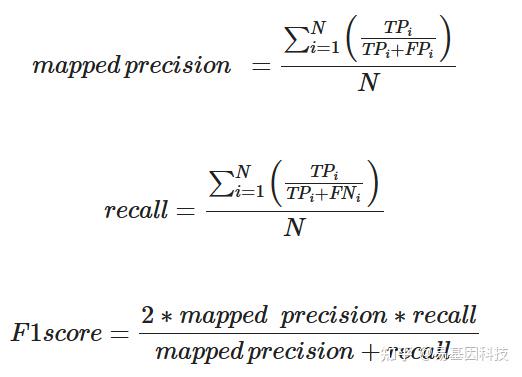
(2)比对软件的唯一比对reads、比对精确率(mapped precision)、召回率(recall)和F1-score
为了研究比对软件的比对性能,利用不同比对软件对模拟数据集A的唯一比对reads、比对精确率、召回率和F1-score进行分析。计算公式如下:
结果发现BSSeeker2-bwt2-2e分别为0、0.25%、0.50%、0.75%和1.00%的测序错误率在三个基因组中表达的唯一比对reads最少,而Bwa-meth表现的最多(图3a)。测序错误率与Hisat_3n_repeat、Hisat_3n、Walt、BSSeeker2-soap2、BSSeeker2-bwt2-local、Batmeth2和BSSeeker2-bwt2-e2e唯一比对的reads数呈高度负相关(Pearson相关系数≤-0.7,P<0.05,图2b)。Bwa-meth、BSBolt、BSMAP、Hisat_3n、Hisat_3n_repeat、Abismal、Bismark-bwt2-e2e和Walt的唯一比对reads率比其他比对软件高22.68%((学生t检验,P<2.2e-16,图3b)。与人物种相比,除了Batmeth2、Hisat_3n_repeat、Bwa-meth和BSBolt外,其他比对软件在牛和猪物种中显示出更高比例的唯一比对reads(图3b)。值得一提的是,BSSeeker2的四种软件软件显示相似的唯一比对reads,但Bismark-bwt2-e2e表现出的唯一比对reads比Bismark-his2多11.00%(图3b)。而在真实数据中没有观察到这些情况(图3c)。

图3:14种比对软件对模拟数据集A和真实数据集A的唯一比对reads、映射精确率、召回率和F1-score分析
模拟数据集A中五个测序错误率的唯一比对reads。
模拟数据集A中这些比对软件的唯一比对reads平均比例。
真实数据集A中这些比对软件的唯一比对reads平均比例。
模拟数据集A中这些比对软件的平均比对精确率和召回率。
模拟数据集A中这些比对软件的平均 F1-score。
模拟数据集 A 中唯一比对reads和F1-score的平均率。
此外,Walt表现出最高的比对精确率和召回率,而BSSeeker2-bwt2-local表现出最低的比对精确率和召回率(图3d)。除了BSSeeker2-bwt外,其他比对软件的比对精确率和召回率与测序错误率呈高度负相关(Pearson相关系数≤-0.7,P<0.05)(图2b)。此外除了BSSeeker2-bwt2-local和Batmeth2外,大多数比对软件在三个基因组中的平均F1-score均>90%,BSSeeker2-bwt2-local的F1-score至少比BSSeeker2-soap2、BSSeeker2-bwt和BSSeeker2-bwt2-e2e低21.64%,而Batmeth2和BSSeeker2-bwt2-local在模拟数据的F1-score上表现较差(图3e),但在真实数据中唯一比对reads上表现出优异的性能(图3c)。三个基因组中的平均唯一比对reads和F1-score结果,显示Bwa-meth、BSBolt、BSMAP、Bismark-bwt2-e2e和Walt的唯一比对reads率和F1-score均>90%(图3f)。因此后续分析的重点将放在Bwa-meth、BSBolt、BSMAP、Bismark-bwt2-e2e和Walt上,以进一步研究其在WGBS数据生物学解释中的表现。
(3)重复序列和CGI对比对性能的影响
由于Bwa-meth、BSBolt、BSMAP、Bismark-bwt2-e2e和Walt在唯一比对reads和F1-score方面表现突出,使用模拟数据集B进一步研究这五种软件的比对性能。结果发现Bwa-meth表现出最少的比对reads数,而Walt表现出最多低质量比对reads(图4a)。测序错误率从0增加到1.00%,BSMAP、BSBolt、Bwa-meth、Bismark-bwt2-e2e和Walt的比对reads分别增加了0.053%、1.95%、0.144%、1.321%和5.651%(图4a)。在测序错误率为0和1.00%的情况下,Bwa-meth的比对reads仅由不正确的唯一比对reads组成(图4a)。与牛和猪物种相比,人物种在五种比对软件中的比对reads更多低质量(P<0.0013,图4a)。且9.2%-53%的低质量比对reads与这五种比对软件中的reads数相同。这一结果反映了这五种比对软件的不同比对性能。

图4:重复序列和CGI对模拟数据集B中5种比对软件比对性能的影响
Bwa-meth、BSBolt、BSMAP、Bismark-bwt2-e2e和Walt的低质量比对reads的比例和类别。
这五种比对软件在重复序列和非重复序列中的低质量比对reads比例。
这五种比对软件在CGI和非CGI中的低质量比对reads比例。

图5:Bwa-meth、BSBolt、BSMAP、Bismark-bwt2-e2e和Walt对真实数据集B中CpG位点和甲基化水平的影响。
Bwa-meth、BSBolt、BSMAP、Bismark-bwt2-e2e和Walt分别检测到的CpG位点数。
通过五种或四种比对软件一致检测到的共同CpG位点数量。
Bwa-meth、BSBolt、BSMAP、Bismark-bwt2-e2e和Walt检测到的准确CpG位点比例。
通过四个或三个比对软件一致检测到的共同CpG位点数量。
Bwa-meth、BSMAP、Bismark-bwt2-e2e和Walt检测到的准确CpG位点比例。
一致和不一致的CpG位点的比例。
CGI、非CGI、重复序列和非重复序列中一致和不一致CpG位点的比例。
一致和不一致CpG比例的高、中、低甲基化水平。
Bwa:Bwa-meth;BSM:BSMAP;Bis:Bismark-bwt2-e2e;Wal:Walt;BSB:BSBolt。
为研究基因组特征对低质量比对reads的影响,分析低质量的比对reads在重复序列和CGI的表征,结果发现在重复序列中表现出比在非重复序列中更低质量的比对reads(学生t-检验,P<2.68e-05,图4b),且在CGI区域中发现比非CGI区域更低质量的比对reads(学生t-检验,P<9.59e-05,图4c)。与非重复序列相比,在Bwa-meth、BSBolt、BSMAP、Bismark-bwt2-e2e和Walt比对软件中,人物种的重复序列低质量比对reads富集度为1.43、1.33、1.41、1.29和1.38,牛物种为4.06、4.29、4.37、2.82和3.03;猪物种为1.70、1.69、1.59、1.50和1.55(Fisher精确检验,P<2.2e-16)(图4b)。此外与非CGI相比(图4c),在Bwa-meth、BSBolt、BSMAP、Bismark-bwt2-e2e和Walt比对软件中,CGI中低质量比对reads在人物种中的富集度为1.67、1.76、1.64、1.67和1.76,牛物种为3.18、2.70、2.23、2.56和2.00,猪物种为1.99、2.06、1.66、1.65和1.69(Fisher精确检验,P<2.2e-16)。这些观察结果表明重复序列和CGI区域导致了低质量的比对reads。
(4)比对软件对CpG位点和甲基化水平的影响
通过Bwa-meth、BSBolt、BSMAP、Bismark-bwt2-e2e和Walt比对软件对真实数据集B 种CpG位点≥10reads覆盖的甲基化谱进行评估,发现BSBolt在人(28551216)、牛(36661360)和猪(31884976)中calling的CpG位点最多;Walt在人(17326301)、牛(20956144)和猪(18884561)中calling的CpG含量最低(图5a)。在人物种中(图5b),这五种比对软件一致检测到8182827个CpG位点,但在去除BSBolt后,Bwa-meth、BSMAP、Bismark-bwt2-e2e和Walt一致检测到的CpG位点增加了170.63%(13962703)。该结果表明BSBolt软件是造成人、牛、猪物种中缺乏一致性CpG位点的原因(图5b)。此外BSMAP在检测CpG位点时表现出最高的准确度(人:15411076,99.37%;牛:14252815,98.99%;猪:14272746,99.03%),而BSBolt的准确度最低(人:9728719,62.73%;牛:6143665,42.67%;猪:6316216,43.82%)(图5C)。由于BSBolt与其他四种软件在CpG位点上存在巨大差异,以下讨论将集中在Bwa-meth、BSMAP、Bismark-bwt2-e2e和Walt上。
就Bwa-meth、BSMAP、Bismark-bwt2-e2e和Walt而言,在去除Walt后,其他三个软件一致地检测到大部分CpG位点,因此可能Walt是导致这四种比对效率不一致的原因(图5d)。BSMAP表现出比其他三种软件更高的精确度(图5e)。通过四种比对软件分别在人、牛和猪中检测到26586835、29736425和28674654个CpG位点,其中约51.65%、42.02%和44.64%被检测为一致CpG位点(图5f)。此外CGI和重复序列在人、牛和猪中可能比非CGI和非重复序列保留更多不一致的CpG位点(学生t-检验,P<6.38e-03,图5g)。在人、牛和猪物种中,一致的CpG在低甲基化(<1/3)和高甲基化(>1/3)时可能过表达,但不一致的CpG可能在中等甲基化时表达下调(1/3~2/3)(学生t检验,P<0.02,图5h)。
(5)比对软件对DMC和DMR的影响
基于真实数据集B中包含≥10 reads覆盖的CpG位点,在Bwa-meth、BSMAP、Bismark-bwt2-e2e和Walt中评估了DMC和DMR的甲基化变化和动态。在这四种软件中,Bwa-meth软件calling的DMC最多(人:1366812;牛:2506659;猪:1747539),Walt软件calling的DMC最少(人:884427;牛:1702832;猪:850291)(图6a)。四种软件在人、牛和猪中一致鉴定出316048、631985和27238个DMC;Bwa-meth、BSMAP和Bismark-bwt2-e2e一致鉴定出最多的DMC(人:432724;牛:811088;猪:363085);Bwa-meth、Bismark-bwt2-e2e和Walt一致鉴定出最少的DMC(人:334856;牛;669034;猪:289236)。在去除Walt后,其他三种比对软件一致鉴定最多DMC,表明Walt对DMC的一致性分析有所欠缺(图6b)。calling DMC时,BSMAP在人(551145,96.70%)、牛(1101408,96.74%)和猪(502505,96.75%)中显示出最高的准确度,而Walt在人(453277,79.53%)、牛(959354,84.27%)和猪(428656,82.54%)中显示出最低的准确度(图6c)。在人、牛和猪的重复序列中,一致性DMC的富集度分别为0.92、0.87和0.83(图6d,Fisher精确检验,P<2.2e-16),表明一致性DMC可能在重复序列中不足,但对CGI没有明显的偏好(图6d)。

图6:Bwa-meth、BSMAP、Bismark-bwt2-e2e和Walt对真实数据集B中的甲基化动态影响
(a) Bwa-meth、BSMAP、Bismark-bwt2-e2e和Walt检测到的DMC数量。
(b) 多种比对软件共同检测到的一致性DMC数量。
(c) Bwa-meth、BSMAP、Bismark-bwt2-e2e和Walt检测到的准确DMC比例。
(d) CGI、非CGI、重复序列和非重复序列中一致和不一致的DMC比例。
(e) Bwa-meth、BSMAP、Bismark-bwt2-e2e和Walt检测到的DMR长度。
(f) 多种比对软件一致检测到的DMR长度。
(g) Bwa-meth、BSMAP、Bismark-bwt2-e2e和Walt检测到的准确DMR比例。
(h) CGI、非CGI、重复序列和非重复序列中一致和不一致的DMR比例。
Bwa:Bwa-meth;BSM:BSMAP;Bis:Bismark-bwt2-e2e;Wal:Walt。
在Bwa-meth、BSMAP、Bismark-bwt2-e2e和Walt中,calling的最长DMR来自Bwa-meth的比对结果(人:109239519 bp,牛:123175355 bp,猪:120202580 bp),calling的最短DMR来自Walt的比对结果(人:77387750 bp,牛:94239118 bp,猪:79180616 bp)(图6e)。Bwa-meth、BSMAP、Bismark-bwt2-e2e和Walt分别在人、牛和猪中共同鉴定出30530480bp、33100297bp和25135110bp的DMR(图6f)。在通过三种软件共同鉴定的DMR中,去除Walt后,Bwa-meth、BSMAP和Bismark-bwt2-e2e共同鉴定的DMR最长(人:40957306 bp;牛:42379448 bp;猪:33330430 bp),表明Walt是缺乏一致性的原因(图6f)。BSMAP在人(50708843 bp,95.39%)、牛(56868993 bp,95.34%)和猪(953420 bp,94.86%)的DMR calling中表现出最高的准确度,而Walt在人(42734834 bp,80.39%)、牛(50368187 bp,84.44%)和猪(39014214 bp,82.64%)中表现出最低的准确度(图6g)。
这些一致性DMR在人、牛和猪的重复序列中的富集分别为0.74、0.74和0.86(图6h,Fisher精确检验,P<2.2e-16),表明一致性DMR也可能在重复序列中不足,且对CGI没有明显的偏好(图6h)。为更好描述四种比对软件对全基因组DNA甲基化谱的影响,观察了SOX9基因在人类、MPZ基因在牛和IGF2BP3基因在猪中的甲基化谱(图7a-c),结果表明比对软件在CpG位点数量和甲基化水平、DMC数量和DMR长度方面生成了不同的甲基化谱。这些结果表明比对软件的选择对甲基化水平具有显著影响。

图7:Bwa-meth、BSMAP、Bismark-bwt2-e2e和Walt揭示基因的DNA甲基化谱
(a) SOX9基因是人对照组和精神分裂症之间差异表达最高的基因。
(b) MPZ基因是与牛胚胎过程Wnt信号通路相关的中枢基因。
(c) IGF2BP3基因是调节猪骨骼肌发育的关键基因。
(6)比对软件对生物解释(biological interpretation)的影响
上述结果表明这四种比对软件的选择对甲基化谱具有显著影响。为了进一步探索比对软件对WGBS数据生物学解释的影响,基于真实数据集B提取DMR相关基因,并通过KEGG富集分析解释和鉴定Bwa-meth、BSMAP、Bismark-bwt2-e2e和Walt比对分析的生物学功能。结果发现Bwa-meth在人(32055)、牛(17762)和猪(22510)中检测到最多的DMR相关基因,而Walt在人(28446)、牛(16841)和猪(20444)中检测到最少的DMR相关基因(图8a)。Bwa-meth、BSMAP、Bismark-bwt2-e2e和Walt在人(25161)、牛(15409)和猪(17967)中共同鉴定出25161个一致性DMR相关基因(图8b)。在通过三种软件共同鉴定的一致性DMR相关基因中,去除Walt,Bwa-meth、BSMAP和Bismark-bwt2-e2e在人(26638)、牛(15893)和猪(18935)鉴定出最多的DMR相关基因(图8b),表明Walt在calling DMR相关基因方面缺乏一致性。对于calling DMR相关基因的准确性,BSMAP在人(27948,98.68%)和牛(16584,99.00%)中最高,Bwa-meth在猪中最高(20011,98.28%),但Walt在人(26846,94.78%)、牛(16267,97.11%)和猪(19392,95.24%)中均最低(图8c)。

图8:Bwa-meth、BSMAP、Bismark-bwt2-e2e和Walt对真实数据集B中生物学解释的影响.
(a) Bwa-meth、BSMAP、Bismark-bwt2-e2e和Walt检测到的DMR相关基因数量。
(b) 通过多种比对软件共同检测到的一致性基因数量。
(c) Bwa-meth、BSMAP、Bismark-bwt2-e2e和Walt检测到的准确DMR相关基因比例。
(d) Bwa-meth、BSMAP、Bismark-bwt2-e2e和Walt检测到的信号通路数量。
(e) 通过多种比对软件共同检测到的一致性通路数。
(f) Bwa-meth、BSMAP、Bismark-bwt2-e2e和Walt检测到的准确通路比例。
Bwa:Bwa-meth;BSM:BSMAP;Bis:Bismark-bwt2-e2e;Wal:Walt。
在DMR相关基因的KEGG分析中,分别在人、牛和猪中鉴定出140、223和223条通路(图8d),通过四种比对软件一致检测出约67、184和157条通路(图8e)。在去除Walt后,其他三种比对软件在人(75)和猪(167)中共同检测到最多的通路;去除Bwa-meth后,其他三种比对软件在牛(189)中共同检测到最多的通路(图8e)。此外,BSMAP在信号通路的calling中表现出最高的准确性(人:86,95.56%;牛:197,99.49%;猪:181,97.84%),Walt在信号通路的calling中表现出最低的准确性(人:82,91.11%;牛:194,97.98%;猪:175,94.59%)(图8f)。
最后分析了四种比对软件中每种软件富集度最高通路的TOP 30。在人物种中,所有四种软件一致地解释了15个(50.00%)信号通路,如粘着斑(Focal adhesion)、轴突导向(Axon guidance)、产热(and Thermogenesis),但其中两种软件可以一致检测到的信号通路数量范围为18~22个(60%-73.33%)。在牛中,所有四种软件都解释了16个(53.33%)信号通路(如MAPK信号通路、肝细胞癌和轴突导向),但四种软件中的两种一致检测到的信号通路数量范围为21~24个(70%-80.00%)。在猪中,所有四种软件都解释了15个(50.00%)信号通路,如内吞作用(Endocytosis)、粘着斑、轴突导向,但四种软件中的两种一致检测到的信号通路数量范围为19~23个(63.33%-76.67%)。
结论
基于14.77 billion reads的真实和模拟WGBS数据,共进行936次比对,以标准化分析评估在人、牛和猪的甲基化研究中的14种常用的比对软件,包括运行时间,内存消耗,唯一比对reads、低质量比对reads、比对精确率、召回率和F1-score,以及在检测CpG位点和甲基化水平、calling DMC/DMR/DMR相关基因和信号通路的生物学解释准确性。
在模拟WGBS数据中,Bwa-meth、BSBolt、BSMAP、Bismark-bwt2-e2e和Walt比其他九种比对软件表现出更高的唯一比对reads,比对精确率、召回率和F1-score。在真实WGBS数据中,与Bwa-meth、BSBolt、Bismark-bwt2-e2e和Walt相比,BSMAP在检测CpG位点和甲基化水平、calling DMC/DMR/DMR相关基因和信号通路方面显示出最高的准确性。这些结果可以为研究人员提供比对软件选择的有用信息,并有助于提高哺乳动物DNA甲基化检测的准确性。
易基因WGBS比对质量评估流程利用具有最高准确性的BSMAP软件进行比对!
关于易基因全基因组重亚硫酸盐测序(WGBS)技术
全基因组重亚硫酸盐甲基化测序(WGBS)可以在全基因组范围内精确的检测所有单个胞嘧啶碱基(C碱基)的甲基化水平,是DNA甲基化研究的金标准。WGBS能为基因组DNA甲基化时空特异性修饰的研究提供重要技术支持,能广泛应用在个体发育、衰老和疾病等生命过程的机制研究中,也是各物种甲基化图谱研究的首选方法。
易基因提供的全基因组甲基化测序技术通过T4-DNA连接酶,在超声波打断基因组DNA片段的两端连接接头序列,连接产物通过重亚硫酸盐处理将未甲基化修饰的胞嘧啶C转变为尿嘧啶U,进而通过接头序列介导的 PCR 技术将尿嘧啶U转变为胸腺嘧啶T。
应用方向:
WGBS广泛用于各种物种,要求全基因组扫描(不错过关键位点)
全基因组甲基化图谱课题
标志物筛选课题
小规模研究课题
技术优势:
应用范围广:适用于所有参考基因组已知物种的甲基化研究;
全基因组覆盖:最大限度地获取完整的全基因组甲基化信息,精确绘制甲基化图谱;
单碱基分辨率:可精确分析每一个C碱基的甲基化状态。
易基因科技提供全面的DNA甲基化研究整体解决方案,技术详情了解请致电易基因。

参考文献:Gong W, et al. Benchmarking DNA methylation analysis of 14 alignment algorithms for whole genome bisulfite sequencing in mammals. Comput Struct Biotechnol J. 2022;20:4704-4716.
相关阅读:
手把手教你做全基因组DNA甲基化测序分析
项目文章|WGBS+RNA-seq揭示PM2.5引起男性生殖障碍的DNA甲基化调控机制
DNA甲基化方法全解析:方法发展、技术应用、优缺点
疾病研究:DNA甲基化与转录组分析联合揭示吸烟免疫相关疾病的表观遗传机制