点击蓝字 关注我们
IPGA原核微生物泛基因组与基因组分析平台

原文链接DOI: https://doi.org/10.1002/imt2.55
研究论文
●2022年9月14日,中国科学院微生物研究所史文聿和马俊才团队在iMeta在线发表了题为“IPGA: A handy integrated prokaryotes genome and pan-genome analysis web service”的文章。
● 本研究提供了一个可以对较大规模微生物基因组进行比较分析的平台IPGA,平台提供了基于基因组注释与泛基因组注释的包括进系统发育分析、基因组共线性分析和核心基因差异分析等后续分析在内的整合流程,并提供了免费、简单的页面操作环境。
● 第一作者:刘冬梅
● 通讯作者:史文聿(shiwy@cau.edu.cn)、马俊才(ma@im.ac.cn)
● 主要单位:中国科学院微生物研究所微生物资源与大数据中心,微生物资源前期开发国家重点实验室;国家微生物数据中心
亮 点
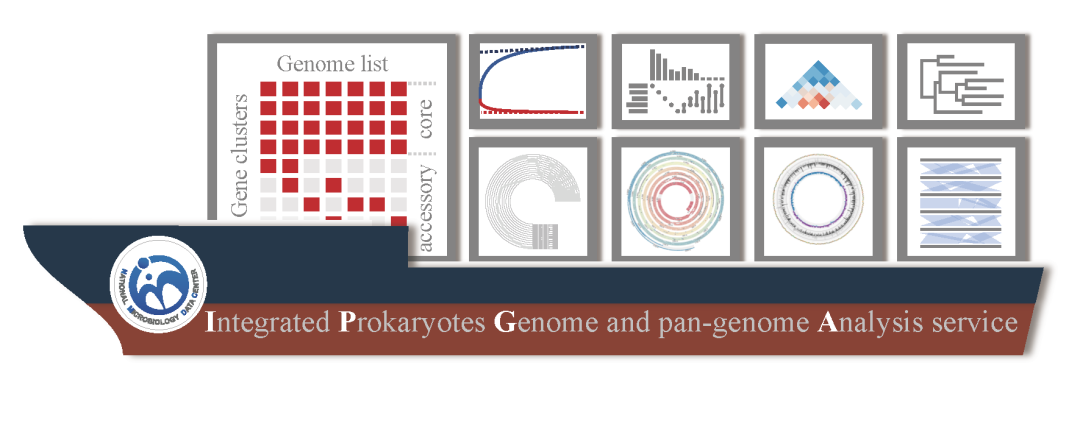
● IPGA是一个免费、易于使用、基于web的在线服务平台,可以为没有生物信息学基础的科研工作人员提供最新的泛基因组分析服务
● IPGA为用户提供了可靠的泛基因组谱,使用户能够进行额外的比较基因组学分析
● IPGA提供了系统发育推断、基因组共线性评估和目标基因组注释等一系列下游分析模块
摘 要
泛基因组研究手段是研究群体基因组的有力的手段之一。虽然目前有很多计算工具来实现这一点,但一个综合的框架来评估它们的性能并为用户提供最佳的选择还从来没有实现过。为了简化大规模原核生物基因组分析的过程,我们引入了集成原核生物基因组和泛基因组分析(IPGA)平台,这是一个一站式的web服务,可以分析、比较和可视化泛基因组以及单个基因组,使用户无需安装任何特定的工具实现该目的。IPGA提出了一个评分系统,帮助用户评估由不同包生成的泛基因组谱的可靠性,并给出最合理的结果,从而用于后续分析。此外,IPGA集成了多个下游比较分析和基因组分析模块,使用户实现多样化的目标。
视频解读
Bilibili:https://www.bilibili.com/video/BV1VG4y1Q7gp/
Youtube:https://youtu.be/N1STFSC7OaA
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
pan-genome中文译作泛基因组,是在研究无乳链球菌中被首次提出,其中的pan代表全部之意(10.1073/pnas.0506758102)。它是用于描述一个类群基因组信息的总和,比单一基因组更能展示出一个类群的基因多样性。从整体范围去研究某一个物种或某一个类群的基因多态性、基因组群体结构、进化趋势比针对单一基因组的研究能够更加清晰的展示背后的生物学逻辑,因此不仅仅局限于微生物,各个物种的泛基因组研究都是在近20年的科学舞台上大放光彩。对于微生物亦是如此,尤其对于致病微生物、工业微生物和模式微生物,泛基因组可以结合其元数据中的表型信息,表征关联的基因组多态性,深入挖掘内在的作用机制,建立可靠的泛基因组参考数据集和数据系统,服务疫情防控、药物设计、工业生产、科学研究。
背 景
泛基因组分析是比较基因组学中最重要的方法之一,广泛应用于原核生物多样性和进化关系的研究、传染病的预防和控制、病原体耐药性的监测等方面。泛基因组分析的目的是评价某一进化支内所有基因和基因组结构的变异性。在泛基因组分析中,最初也是最重要的一步是同源基因的聚类。基因簇根据它们在给定基因组集中的出现情况分为三组:核心基因、辅助基因和独特基因。目前,有许多软件包或web服务可以实现这一目标,但泛基因组分析的结果可能会非常不同。因此,评估该结果的可靠性比较重要。此外,提供用户所需要的目标分支或目标菌株的下游分析和可视化也会极大限制非生物信息学家对原核生物泛基因组的研究。
因此,我们提供了IPGA,一个强大而实用的在线服务平台,能够实现上述目标并可实现下游满足不同目的的多种分析需求。IPGA集成了8个泛基因组分析包,并提供了一个评估策略,帮助用户选择最佳的泛基因组图谱。此外,IPGA还允许用户同时执行系统发育推断、基因组共线性评估和目标基因组注释等下游分析。
实 现
IPGA是基于国家微生物数据中心(NMDC)云系统开发的,为IPGA的业务提供了充足的计算资源。这里面,动态调度系统是由Mesos和Marathon框架来完成的,封装在IPGA中的所有软件包都被单独封装,并由调度系统启动。网站前端采用spring cloud框架和Java SE Development Kit 8开发。此外,IPGA可以通过NMDC系统中基因组数据链接API获取到所有基因组数据。因此,用户只需为已有的公开的基因组提供基因组accession ID,而无需上传基因组文件,便可以方便使用它们。
IPGA工作流接受一组原核基因组作为输入。然后,可以选择7个分析模块启动IPGA工作流。最初启动的是质量控制模块,用于去除所有低质量基因组,并对每个基因组进行物种分类识别。然后IPGA预测所有筛选后基因组的基因,并将其作为泛基因组分析模块的输入。用不同的软件创建泛基因组谱后,IPGA提取每对属于同一核心基因簇的基因之间的所有链接,然后统计不同软件标注的不同泛基因组谱之间共享的链接数。IPGA将基于COG注释和多个软件投票的方式确定哪些链接应该被加分,从而计算出每个泛基因组谱得分,最高分的结果将会被汇报给用户。在此之后,筛选的基因组和基因簇将用于接下来的四个下游比较基因组分析模块,包括系统发育分析模块、基因组共线性分析模块、核心基因等位基因分析模块和平均核苷酸一致性(ANI)计算模块。此外,可以在基因组注释模块中选择要注释的基因组列表。在执行流程中,根据用户的需求,任何模块未被选中都可以跳过。上传所有输入基因组序列并确定分析过程和相关的所有参数后,可以启动IPGA工作流程。作业状态和结果将通过电子邮件发送给作业提交者。
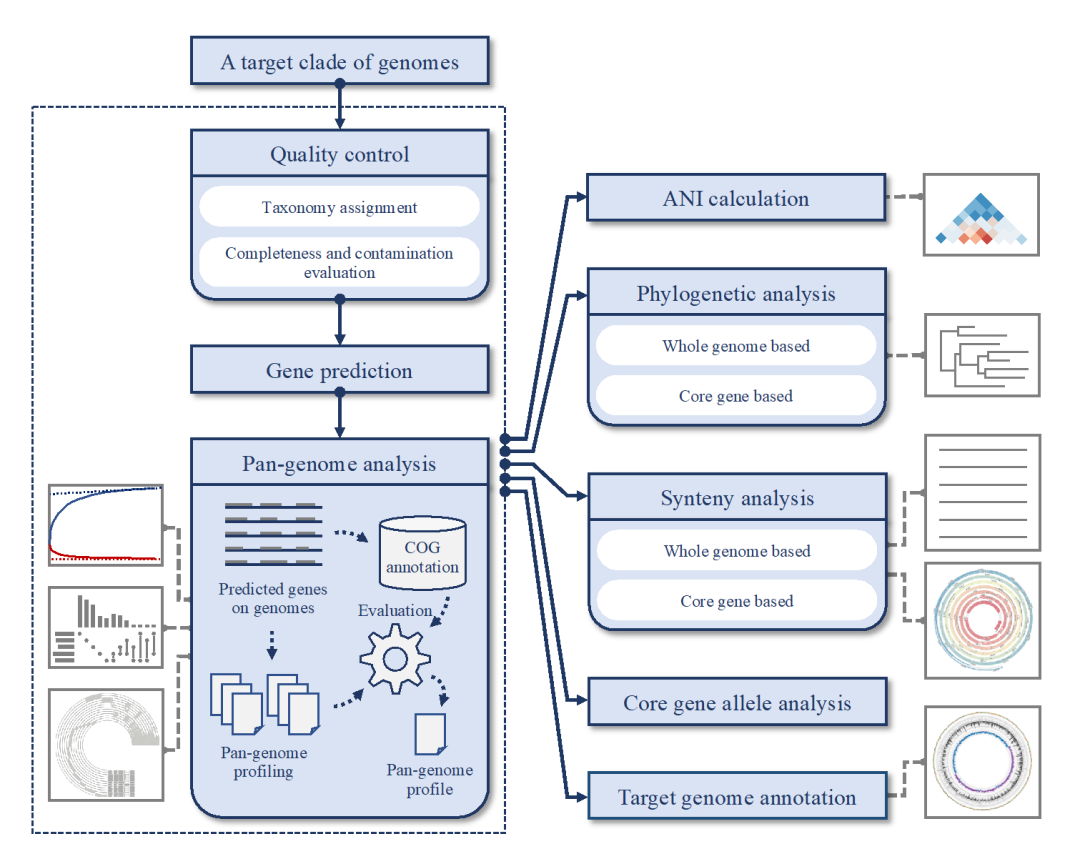
图1. IPGA的工作流程
IPGA,综合原核生物基因组和泛基因组分析。
结 果
文中展示了9个不同的数据集演示了IPGA泛基因组分析模块的性能,包括宏基因组组装的基因组,食源性病原体的基因组,以及来自几种细菌分支的基因组。8种软件的泛基因组分析结果在这些测试数据集之间存在很大差异(图2A)。在数据集Capnocytophaga和Cellulosilyticum的泛基因组图谱中,IPGA提取了分组在同一簇中的基因对,然后,统计不同泛基因组谱中相同基因对的数量。Roary、panX、OrthoFinder和PPanGGoLiN共享的基因对最多,在这两个数据集中表现也较为可靠(图2B)。从对所有数据集的评价过程结果来看,OrthoFinder、panX和PPanGGoLiN的性能相对稳定,Roary低估了所有数据集的核心基因簇数量,尤其是在大数据集(>200个基因组)中。与其他泛基因组分析平台相比,IPGA提供了性能评估,并提供了测量的泛基因组剖面来进行下游分析。
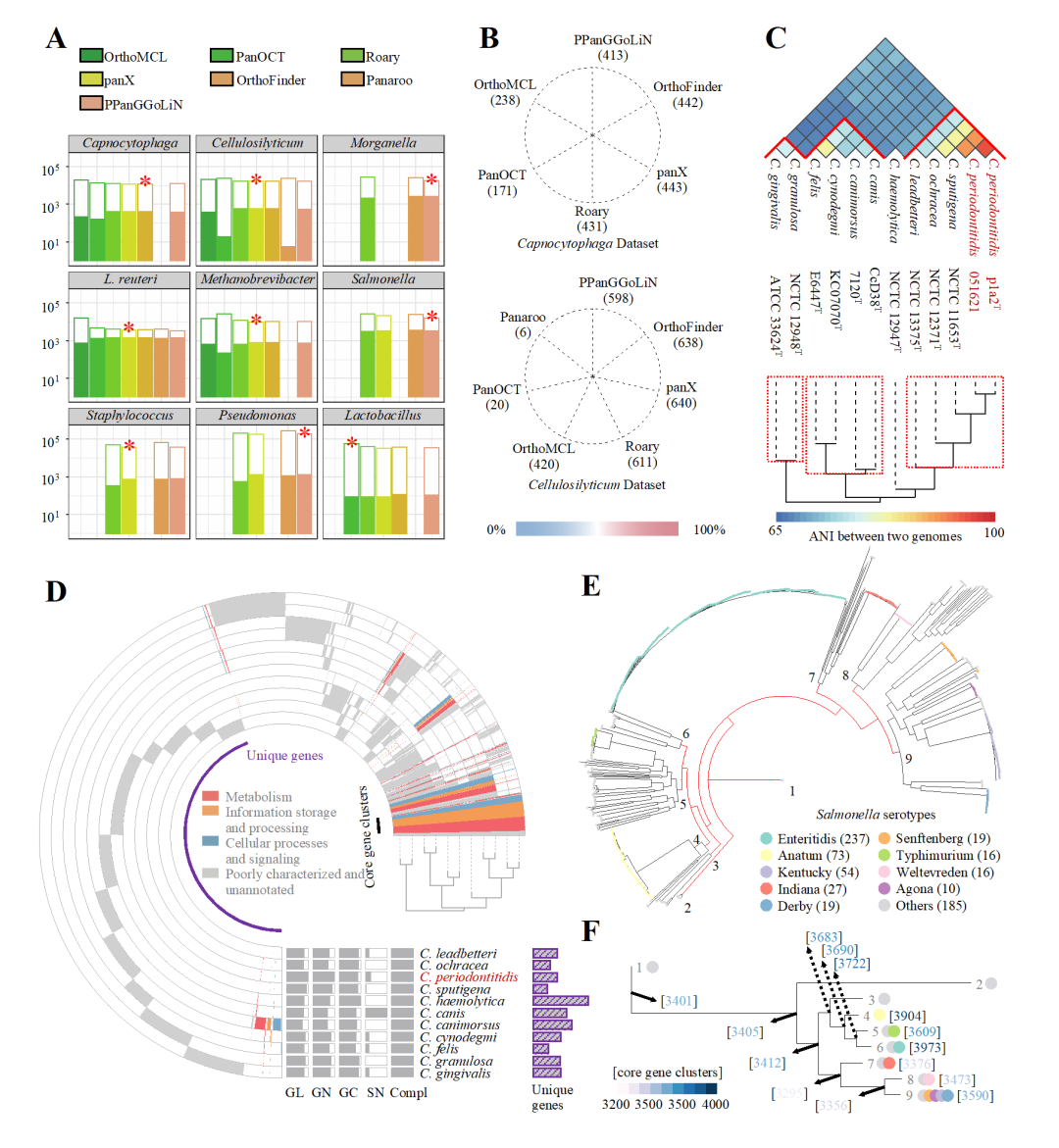
图2. IPGA在不同数据集上的结果
(A) 对9个不同数据集的性能评估。条形图的高度代表每个工具估计的总的正交基因簇的数量,彩色部分代表每个工具预测的核心基因的数量。工具是用颜色编码的,每个数据集的最佳工具用星号标记。(B) 4个不同的软件之间共享的核心基因链接的数量。(C) 数据集Capnocytophaga的ANI分析结果。(D) Capnocytophaga属的所有类型菌株的泛基因组概况。"GL"、"GN"、"GC"、"SN "和 "Compl "分别代表基因组长度、基因数、GC含量、支架数和基因组完整度。(E)基于数据集沙门氏菌的全基因组变异的系统发育推断。(F)数据集沙门氏菌九个主要聚类的核心基因聚类。ANI,平均核苷酸身份;IPGA,综合原核生物基因组和泛基因组分析。
IPGA应用举例一
新物种的提出建议采用泛基因组分析来提供新物种与已发表物种的基因组层面的差异性。我们使用数据集Capnocytophaga演示了一个案例研究,其中包含属于11个物种的12个基因组。在这些基因组中,C. periodontitidis作为新分类菌株被提出。根据ANI值(cutoff = 75,图2C)可将该属11种全部分为4个类群。在11716个同源基因簇中,核心基因簇仅有419个(3.57%),而不同基因组的独有基因簇多达6976个(59.54%)。其中,C. periodontitidis (559个)的独特基因多于C. ochracea(398个)、C. sputigena(273个)和C. felis(356个)的独有基因(图2D)。这些结果都提供了基因水平的证据,表明其与Capnocytophaga属的其他模式菌有很大的差异。
IPGA应用举例二
环形基因组图是基因组注释的常用方法。然而,使用循环基因组表示来可视化碎片化的基因组,特别是对于来自宏基因组数据的高质量组装基因组,具有误导性。我们展示了Cellulosilyticum属的两个基因组,一个来自分离菌株(菌株WCF-2)的完整基因组,使用CGView来展示(下图A);另一个宏基因组组装基因组(MAG SIG270),使用IPGA绘制螺线图(下图B)。两条链所有基因的gc-skew、gc-content和基因功能注释均显示在不同的环的通道上。
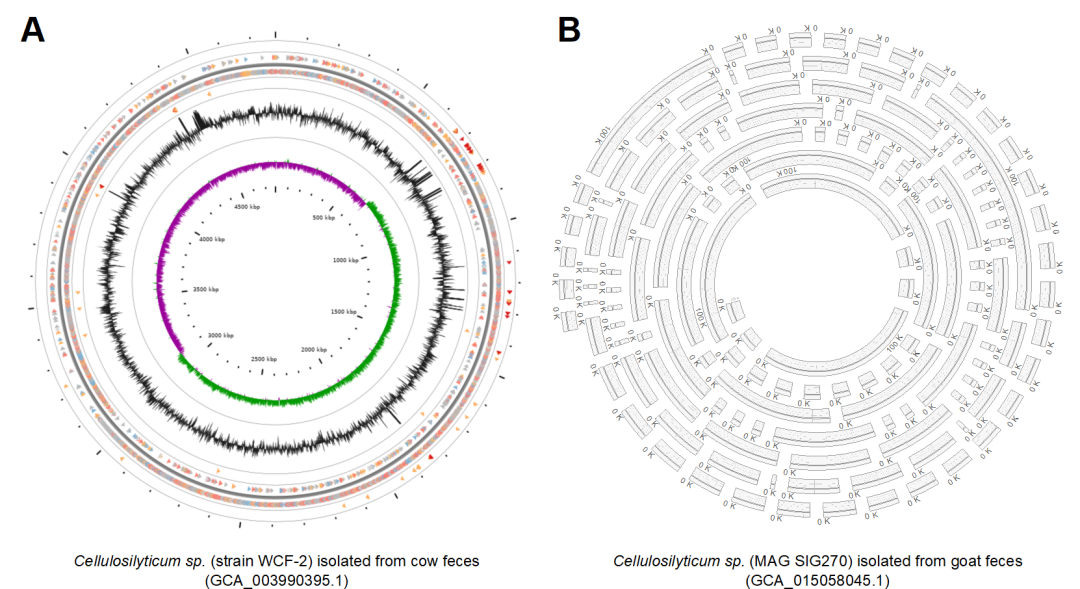
IPGA应用举例三
IPGA还可以帮助用户在大量基因组的研究中进行下游分析。以enterobase中标记为从中国分离得到的667个高质量沙门氏菌基因组的数据集为例。基于全基因组SNP的系统发育推断,这些基因组可以分为9个不同的簇(图2E)。由于其中有12个基因组与其他基因组之间的ANI值较低,并且属于一个亚分支,在后面展示中,这个极长的分支被移除(下图左),泛基因组图谱如图2F所示。因此,用户可以根据详细的输出,关注目标簇的基因差异或单核苷酸差异。除了标准的系统发育分析,我们还展示了另一个使用乳酸杆菌属的131个完整基因组的例子。用户可以放大一个分支,以发现一个亚分支中的八个基因组、七个L. iners基因组和一个外组基因组之间的基因组共线性分析结果(下图右)。
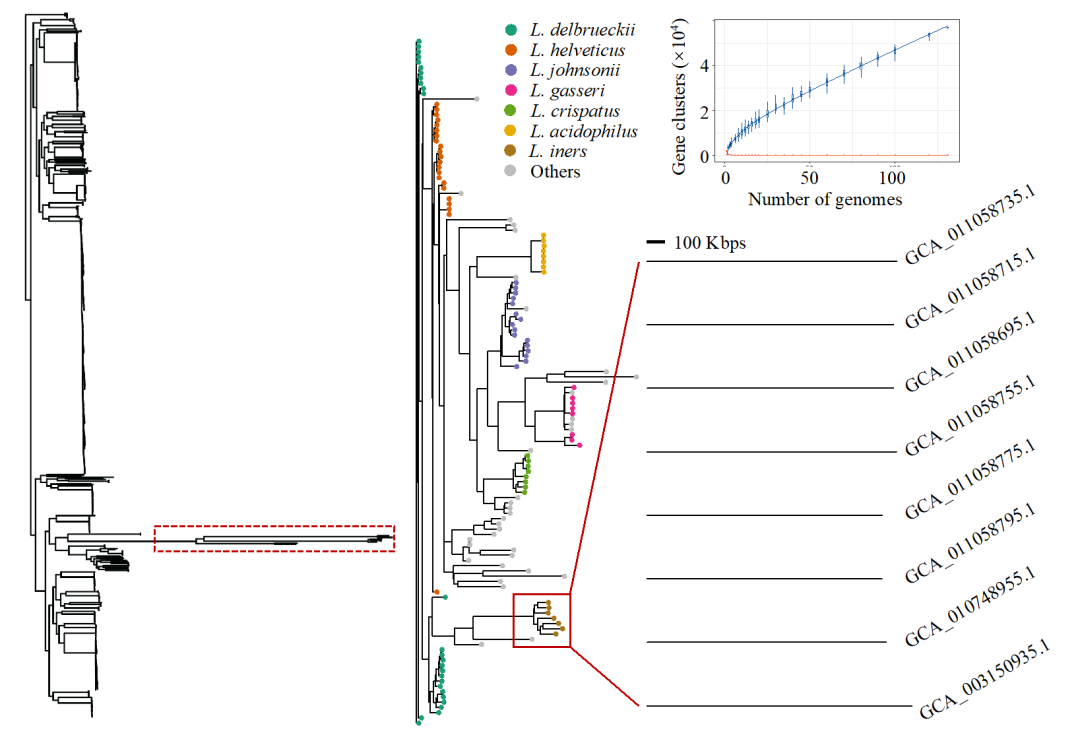
平台链接
● 平台入口:https://nmdc.cn/ipga/
● 使用说明:https://nmdc.cn/ipga/ => 子页面manual
● 视频教程:https://nmdc.cn/ipga/ => 子页面video
引文格式:
Dongmei Liu, Yifei Zhang, Guomei Fan, Dingzhong Sun, Xingjiao Zhang, Zhengfei Yu, Jinfeng Wang, Linhuan Wu, Wenyu Shi, Juncai Ma. 2022. IPGA: a handy integrated prokaryotes genome and pan-genome analysis web service. iMeta 1: e55. https://doi.org/10.1002/imt2.55
作者简介
国家微生物科学数据中心(https://nmdc.cn/)于2019年以中国科学院微生物研究所作为依托单位建立,承担微生物科学数据的汇交管理、资源挖掘、数据应用等业务内容,中心数据资源总量超过2PB,数据记录数超过40亿条,覆盖微生物资源检索信息、微生物研究项目信息、微生物及微生物组原始测序数据、微生物多组学数据以及微生物相关文献专利等微生物研究的全生命周期。数据中心开发了一系列数据平台,重点推进微生物领域科技资源向国家平台汇聚与整合,加强微生物资源开发应用与分析挖掘,提升微生物资源有效利用和科技创新支撑能力,为科学研究、技术进步和社会发展提供高质量的科技资源共享服务。

史文聿(通讯作者)
● 中国科学院微生物研究所助理研究员
● 从事大数据解析微生物基因组特征、功能组学平台设计与优化、微生物与微生物组人工智能算法设计等研究。在iMeta、mLife、Nucleic Acids Research和Gut等刊物以第一和共同第一发表多篇文章

马俊才(通讯作者)
● 中国科学院微生物研究所研究员,国家微生物科学数据中心主任,世界微生物数据中心主任
● 主持51个国家150个机构参加的国际数据合作和全球万种微生物模式菌株测序计划,主持制定国际微生物领域第一个ISO级别的数据标准。在iMeta,Nucleic Acids Research,GigaScience,BMC genomics等刊物以通讯作者发表多篇文章
更多推荐
(▼ 点击跳转)
高引文章 ▸▸▸▸
iMeta | 德国国家肿瘤中心顾祖光发表复杂热图(ComplexHeatmap)可视化方法
▸▸▸▸
iMeta | 浙大倪艳组MetOrigin实现代谢物溯源和肠道微生物组与代谢组整合分析
▸▸▸▸
iMeta | 高颜值绘图网站imageGP+视频教程合集

第1卷第1期

第1卷第2期

第1卷第3期

第1卷第4期
期刊简介
“iMeta” 是由威立、肠菌分会和本领域数百位华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表原创研究、方法和综述以促进宏基因组学、微生物组和生物信息学发展。目标是发表前10%(IF > 15)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、前3年免出版费、50万用户的社交媒体宣传等。2022年2月正式创刊发行!
联系我们
iMeta主页:http://www.imeta.science
出版社:https://onlinelibrary.wiley.com/journal/2770596x
投稿:https://mc.manuscriptcentral.com/imeta
邮箱:office@imeta.science
往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|




























机器学习
后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集