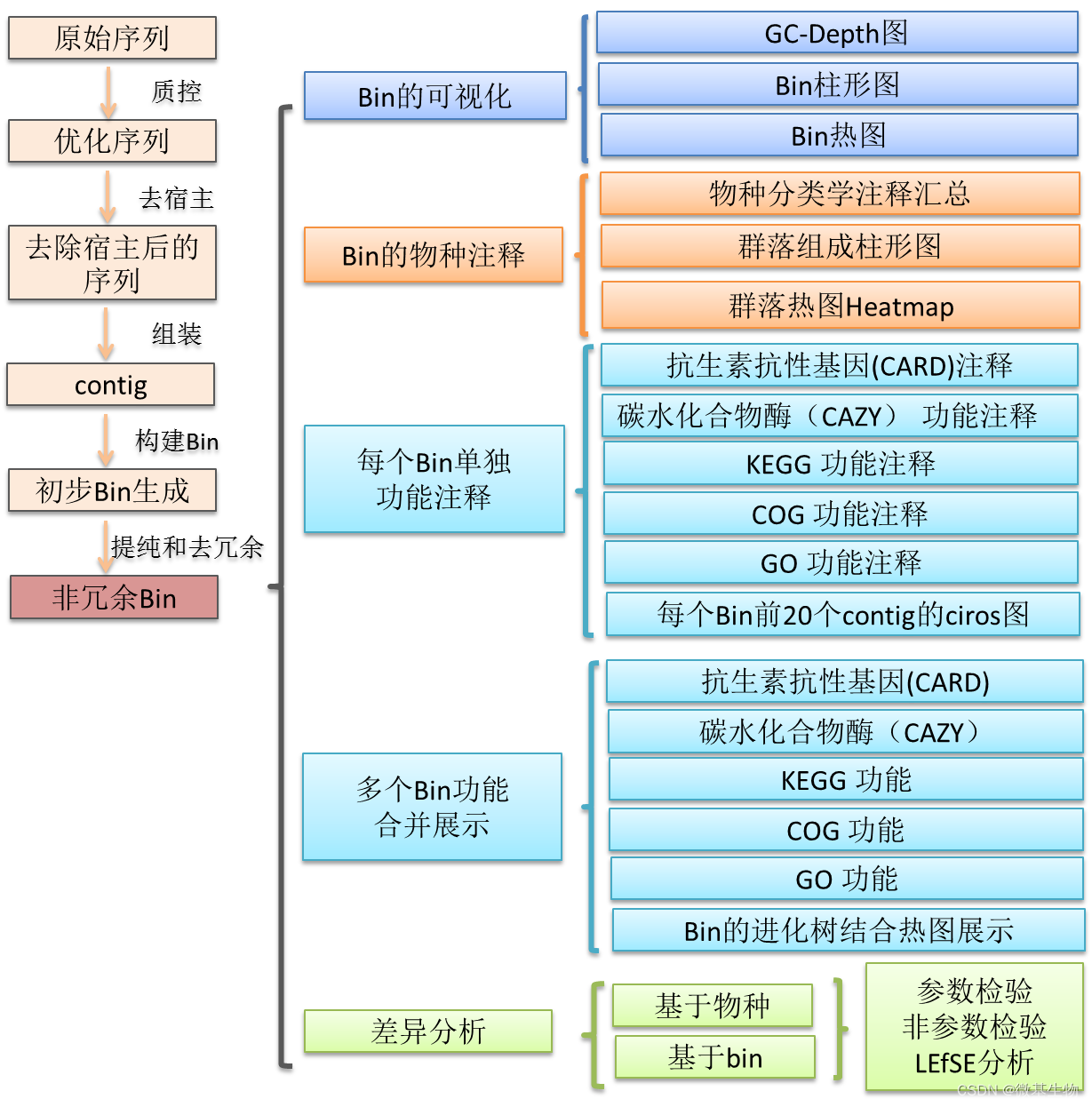
一、介绍
宏基因组 ( Metagenome) 指特定环境下所有生物遗传物质的总和。它包含了可培养的和未可培养的微生物的基因。一般从环境样品中提取基因组DNA, 进行高通量测序,从而分析微生物多样性、种群结构、功能信息、与环境之间的关系等。
宏基因组的分析目前主要包括三种方法:基于组装分析、基于reads分析、基于bin分析。
下面我们介绍基于组装的分析方法。
二、分析流程介绍
数据分析从下机原始序列开始,首先对原始序列进行去接头、 质量剪切以及去除污染等优化处理。然后使用优质序列进行拼接组装和基因预测,将各样本预测得到的基因集合并在一起去冗余,得到非冗余基因集;对得到的非冗余基因集进行物种和功能上的注释,并使用BWA软件将优化序列比对到非冗余基因集,计算得到各基因在各样品中的丰度信息(RPKM); 对物种和功能注释结果进行统计分析。
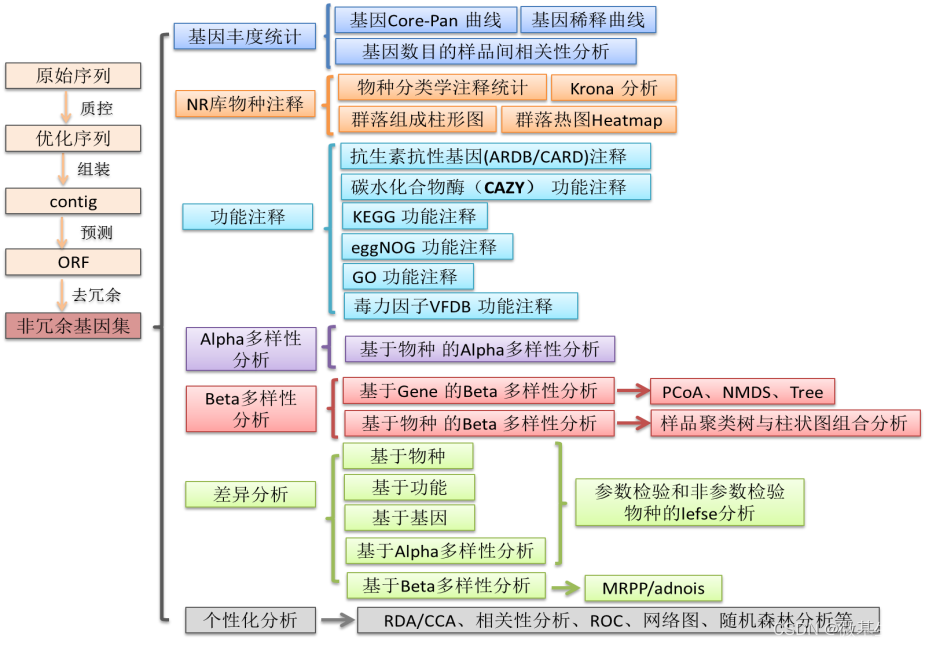
三、详细流程
- 使用fastp软件使用划框方法去除低质量碱基,同时去除接头序列;如果样品来源于宿主(比如人或动物的粪便),而且该宿主本身的基因组已被发表, 则通过软件Bowtie2将reads比对宿主DNA序列,并去除比对相似性高的污染reads;
- MEGAHIT是一个二代测序从头组装工具,尤其在土壤等复杂环境样本组装、大量样本混合组装方面优势明显,同时提供更好的完整性和连续性,为行业的主流组装软件。使用Megahit软件通过设置不同kmer参数,对优化序列进行组装得到Contigs;可以通过N50判断组装结果的质量。
| 样品名称 | 序列数(条) | 碱基数(bp) | Mean(bp) | Max(bp) | N50(bp) | N70(bp) | N90(bp) |
| Sample1 | 118571 | 2.47E+08 | 2079 | 736220 | 13365 | 2240 | 598 |
| Sample2 | 160636 | 2.74E+08 | 1703 | 764995 | 7111 | 1569 | 513 |
| Sample3 | 120704 | 2.23E+08 | 1845 | 627035 | 10082 | 1827 | 536 |
- Prodigal是一种采用无监督的机器学习算法,用于原核基因组的蛋白质编码基因预测软件工具。我们使用Prodigal软件对Contigs进行基因预测(ORF);
- 使用CD-Hit软件对基因集去冗余,得到非冗余基因集 (non-redundant gene catalog);
- 基因丰度统计:使⽤BWA软件,将质控优化序列⽐对到非冗余基因集上,从比对上的 序列数目及基因长度出发,计算得到各基因在各样品中的丰度信息(RPKM);
- 使用DIAMOND软件将非冗余基因集与 NCBI 的 NR序列进行blastp比对,得到基因序列对应的物种注释信息;
- 使用DIAMOND软件将非冗余基因集与ARDB、CAZy、KEGG、eggNOG、VFDB等数据库进行blastp比对,得到基因序列对应的功能信息。
四、主要结果介绍
1、基于基因的分析
1.1 样品间相关性分析
生物学重复是任何生物学实验所必须的,高通量测序技术也不例外。 样品间基因丰度相关性是检验实验可靠性和样本选择是否合理性的重要指标。 相关系数越接近1,表明样品之间基因丰度模式的相似度越高。
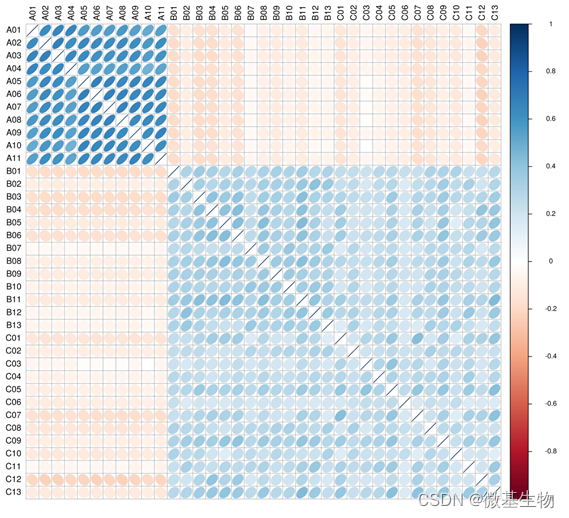
注:图中不同颜色代表 spearman 相关系数的高低; 相关系数与颜色间的关系见右侧图例说明; 颜色越深代表样品间相关系数的绝对值越大; 椭圆向左偏表明相关系数为正,右偏为负; 椭圆越扁说明相关系数的绝对值越大。
2、基于物种的分析
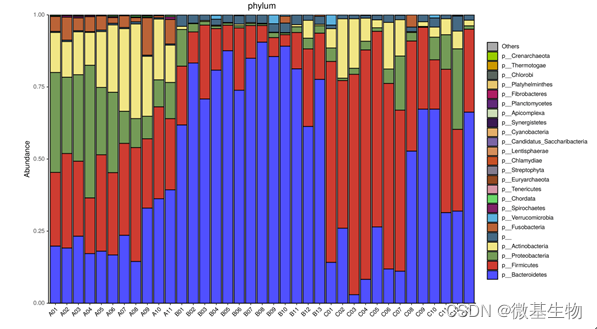
从不同分类层级(门纲目科属种)的相对丰度表出发,展示相对丰度前25的物种,其余的合并为Others,绘制出各样品对应的物种注释结果在不同分类层级上的相对丰度柱形图。
注:横坐标为样本,纵坐标为各级别的相对丰度;其它级别的图请在下面目录查看。
3、功能分析
抗生素抗性基因数据库(ARDB,ARDB-Antibiotic Resistance Genes Database) 收集了不同环境来源的(如肠道、生活废水、河流等)细菌抗药性 基因及其抗性谱、作用机制、本体论、COG和CDD等注释信息,为研 究药物作用、环境治理提供研究依据。
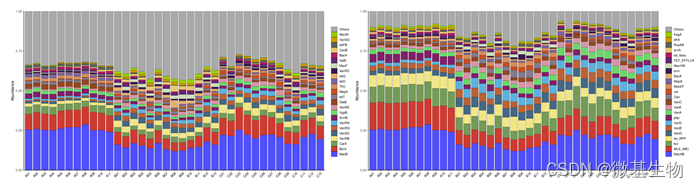
抗性基因类型统计(Type 左) (Class 右)
The Comprehensive Antibiotic Resistance Database (http://arpcard.mcmaster.ca)CARD 数据库核心是 ARO(Antibiotic Resistance Ontology),ARO 包含了与抗生素抗性基因,抗性机制,抗生素和靶相关的term。CARD 数据库已成为目前最受欢迎的耐药基因研究工具之一。
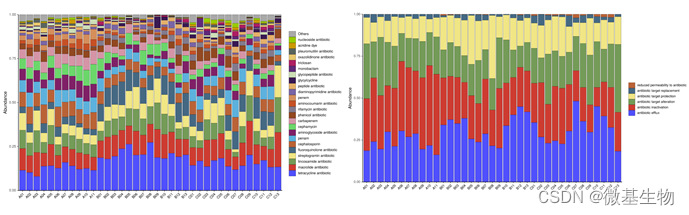
抗性基因统计(Drug_Class左) (Resistance_Mechanism右)
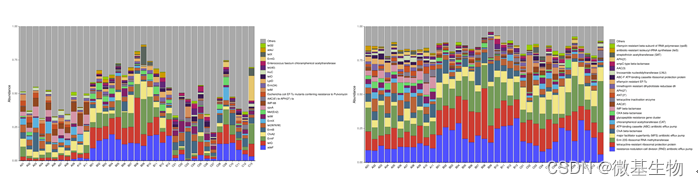
抗性基因统计(Best_Hit_ARO左) (AMR_Gene_Family 右)
3.2 碳水化合物酶功能注释(CAZY)
碳水化合物活性酶(Carbohydrate-active enzymes,CAZyme) 对地球上所有碳水化合物的合成、降解与修饰起重要作用, 因此深入研究CAZyme,对于了解微生物碳水化合物的代谢机制非常重要。
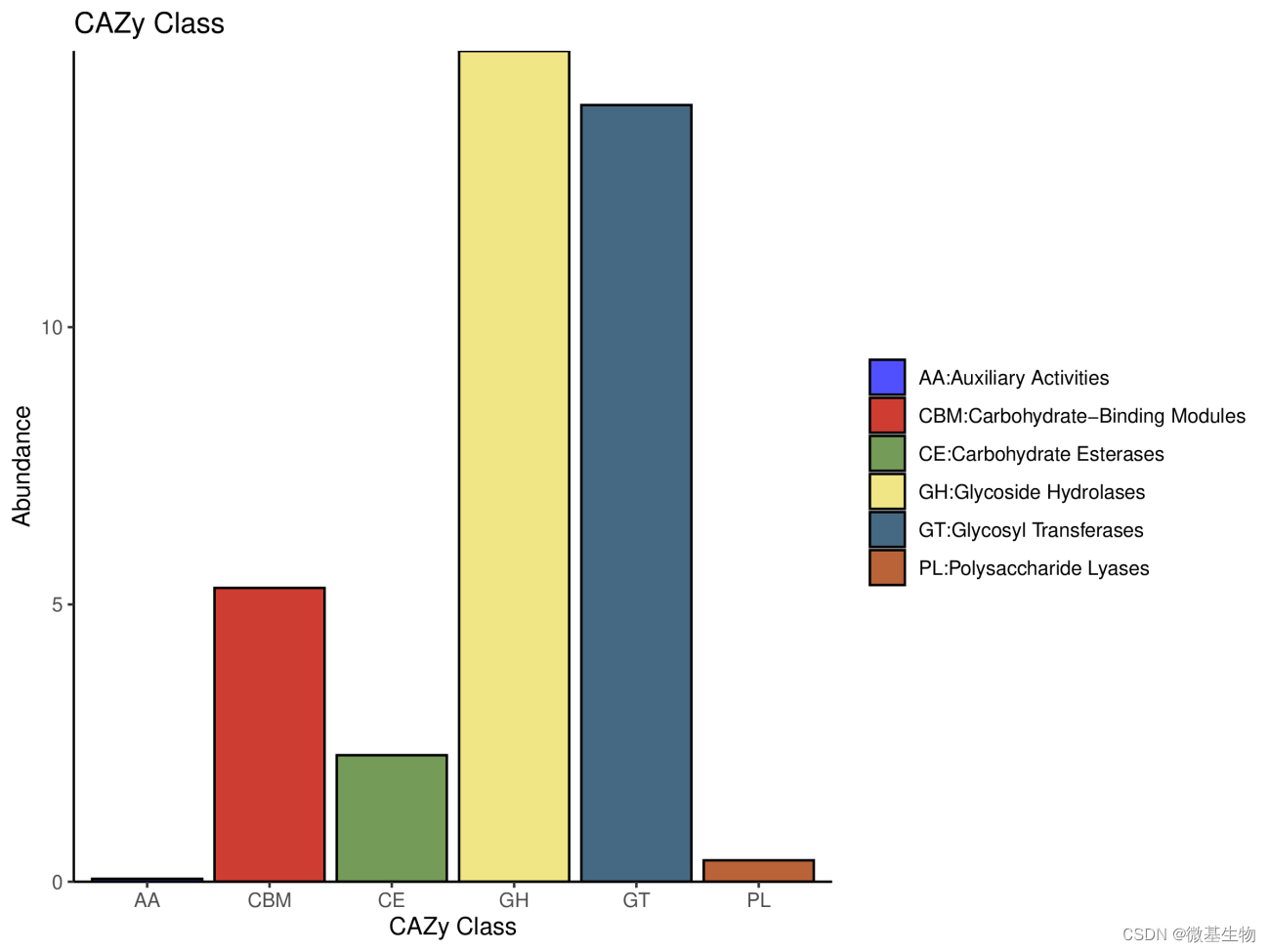
注: 横坐标6大功能类,纵坐标基因丰度,颜色表示功能。
KEGG数据库(Kyoto Encyclopedia of Genes and Genomes, 京都基因和基因组百科全书,KEGG: Kyoto Encyclopedia of Genes and Genomes) 是系统分析基因功能,联系基因组信息和功能信息的大型知识库。 KEGG GENES数据库提供关于在基因组计划中发现的基因和蛋白质的序列信息; KEGG PATHWAY数据库包括各种代谢通路、合成通路、膜转运、信号传递、 细胞周期以及疾病相关通路等。
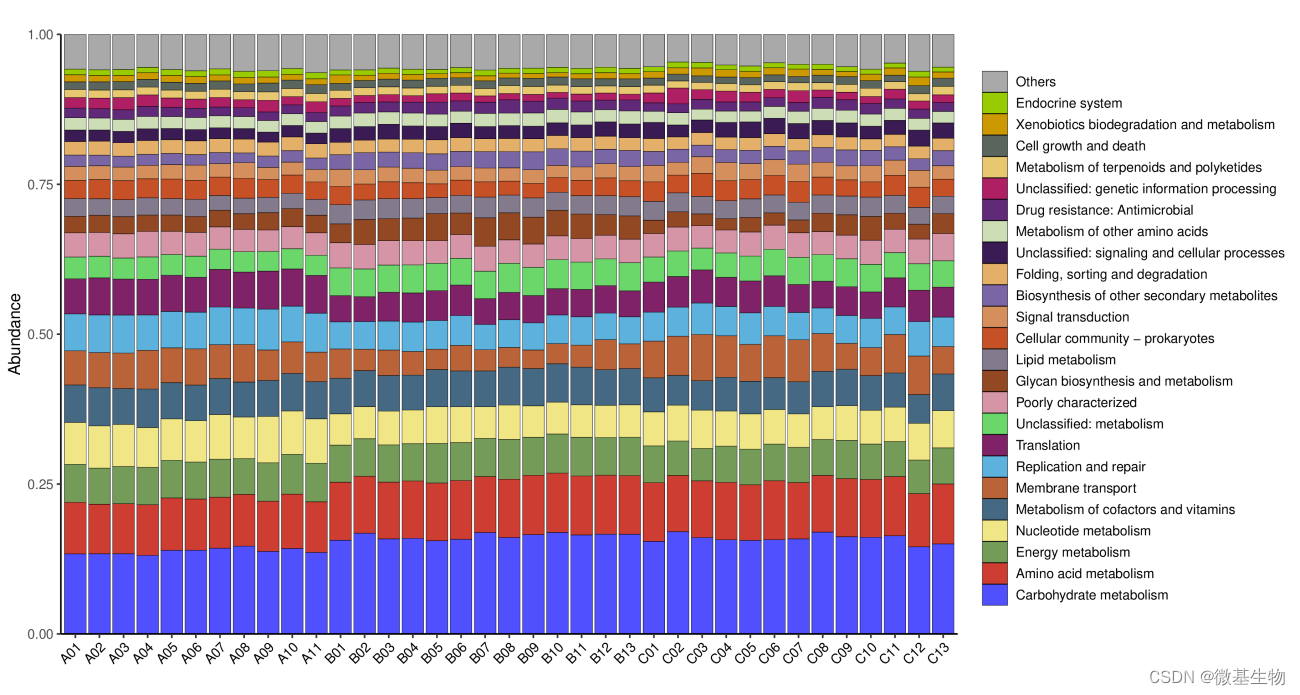
注: 横坐标每个样本,纵坐标基因丰度,颜色表示名称。
3.4 eggNOG功能注释
eggNOG 数据库:是国际上普遍认可的同源聚类基因群的专业注释数据库,包括来自原始COG/KOG的功能分类,以及基于分类学的功能注释。
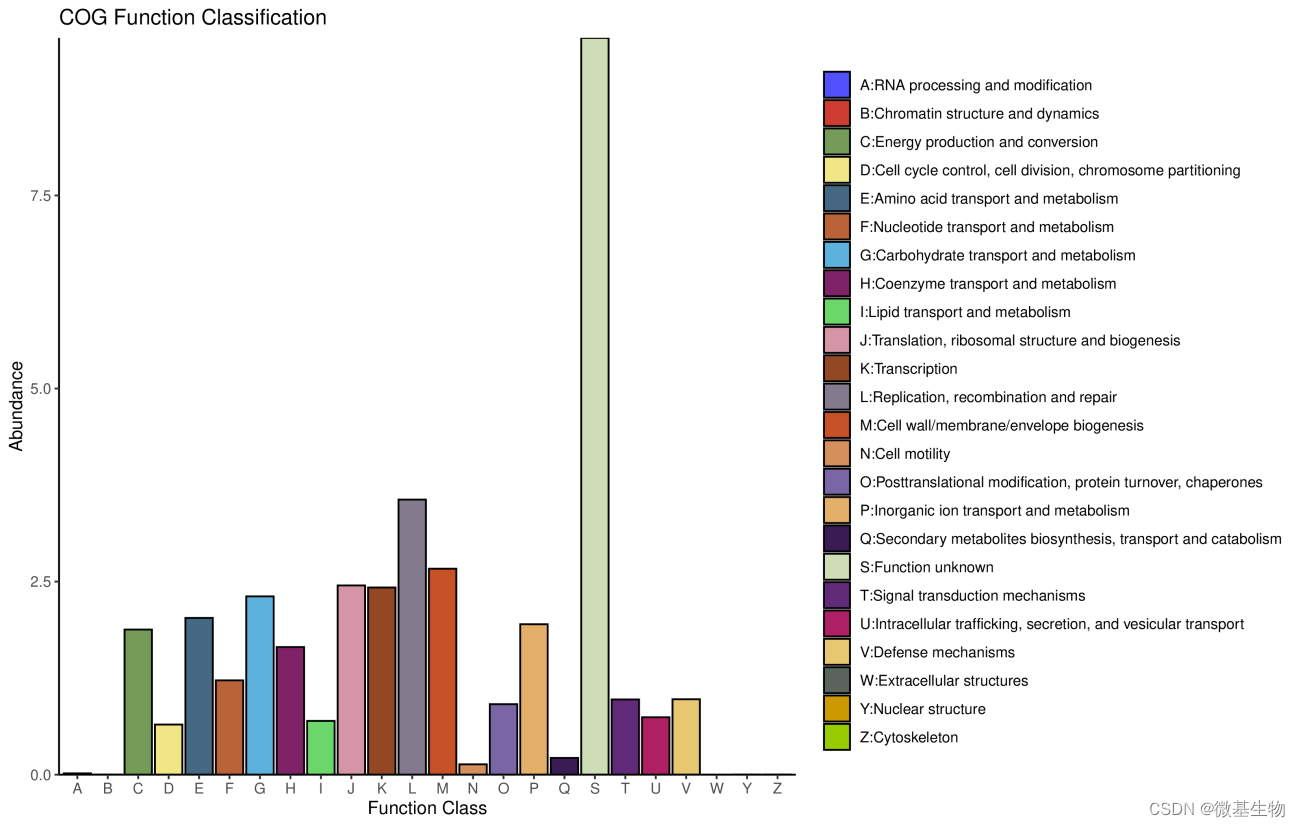
注: 横坐标每个功能,纵坐标基因丰度,颜色表示每个功能。
GO数据库分别从功能、参与的生物途径及细胞中的定位对基因产物进行了标准化描述,所谓的 GO,是生物学功能注释的一个标准词汇表术语(GO term),将基因的功能分为三部分: 基因执行的分子功能( Molecular Function), 基因参与的生物学过程(Biological Process), 基因所处的细胞组分( Cellular Component)。

注: 横坐标表示 Level2 水平上 GO term,不同颜色代表不同 GO 类,分为三大类,纵坐标表示 注释到该 term 上面的 Unigene 数目
4 Alpha多样性:
Alpha 多样性( Alphadiversity)包括 :
chao 指数和 ACE 指数反映样品中群落的丰富度( speciesrichness),即简单指群落中物种的数量,而不考虑群落中每个物种的丰度情况。
shannon 指数以及 simpson 指数反映群落的多样性( speciesdiversity),受样品群落中物种丰富度( speciesrichness)和物种均匀度( speciesevenness)的影响。
相同物种丰富度的情况下,群落中各物种具有越大的均匀度,则认为群落具有越大的多样性。
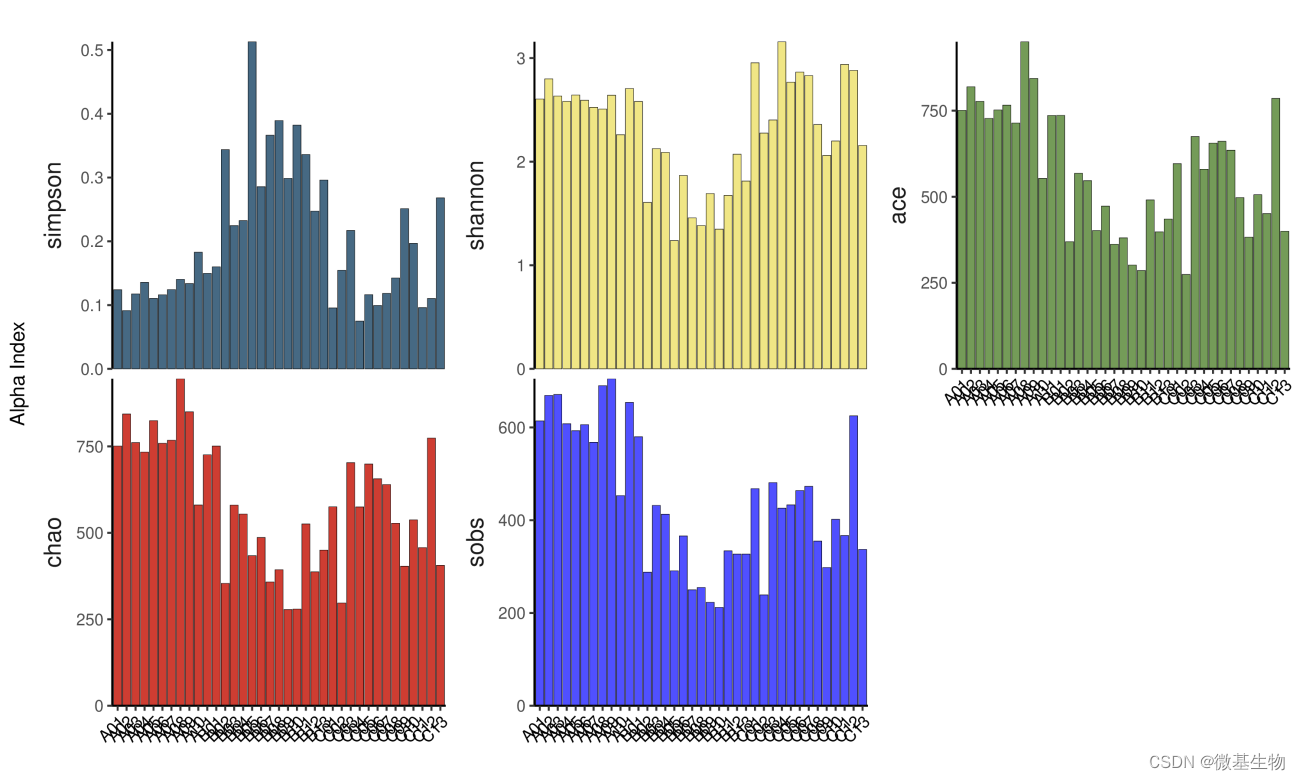
5 Beta多样性分析
5.1 基于基因分析
Beta多样性(Beta diversity)分析是用来比较一对样品在物种多样性方面存在的差异大小。分析各类群在样品中的含量,进而计算出不同样品间的Beta多样性值。多种指数可以衡量Beta多样性,常用的为Bray-Curtis,Jaccard。我们将以这两种方法得到的矩阵,做NMDS,PCoA及聚类树分析。
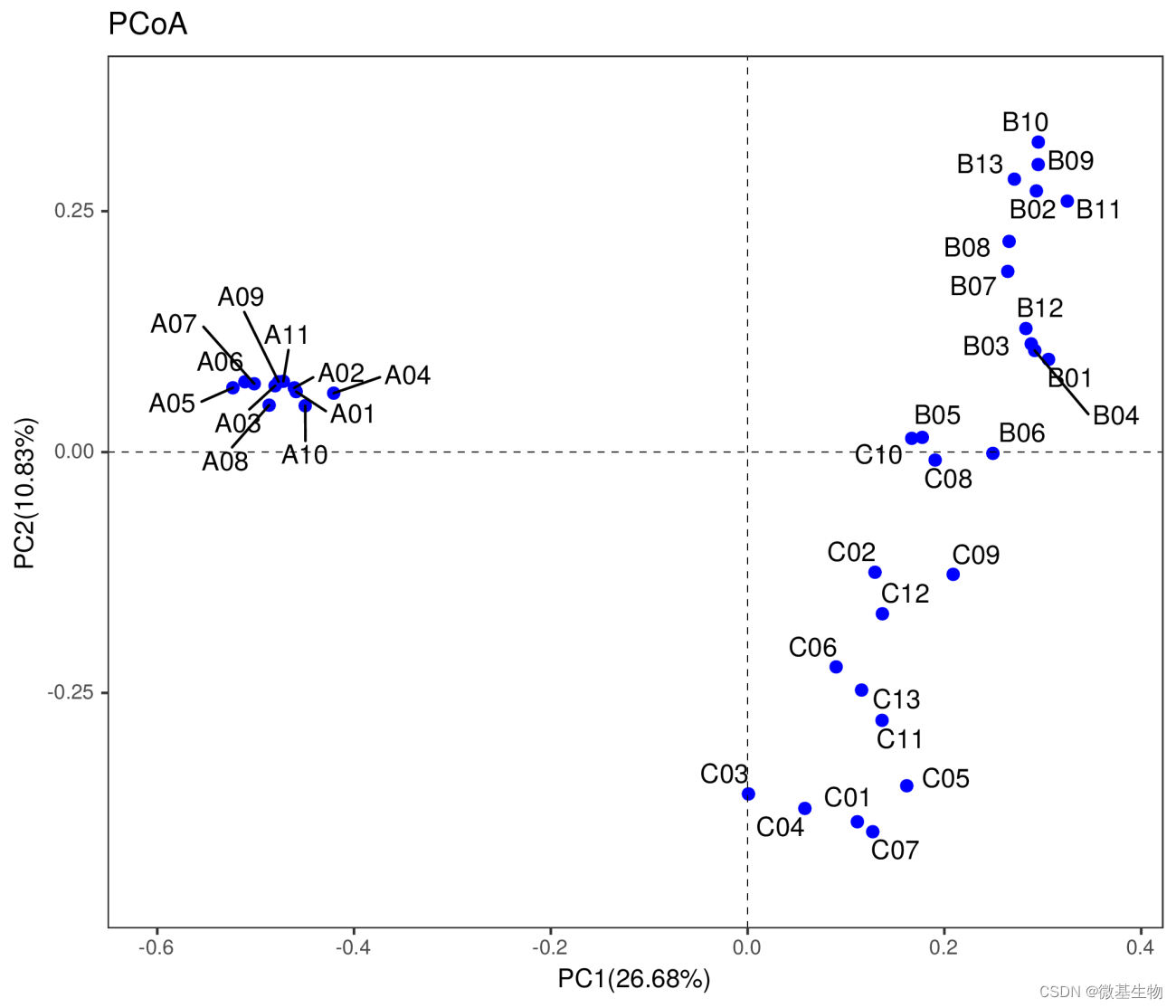
5.2 基于物种分析
左边是样品间基于群落组成的层次聚类分析(Bray-curtis 算法),右边是样品的群落结构柱状图。
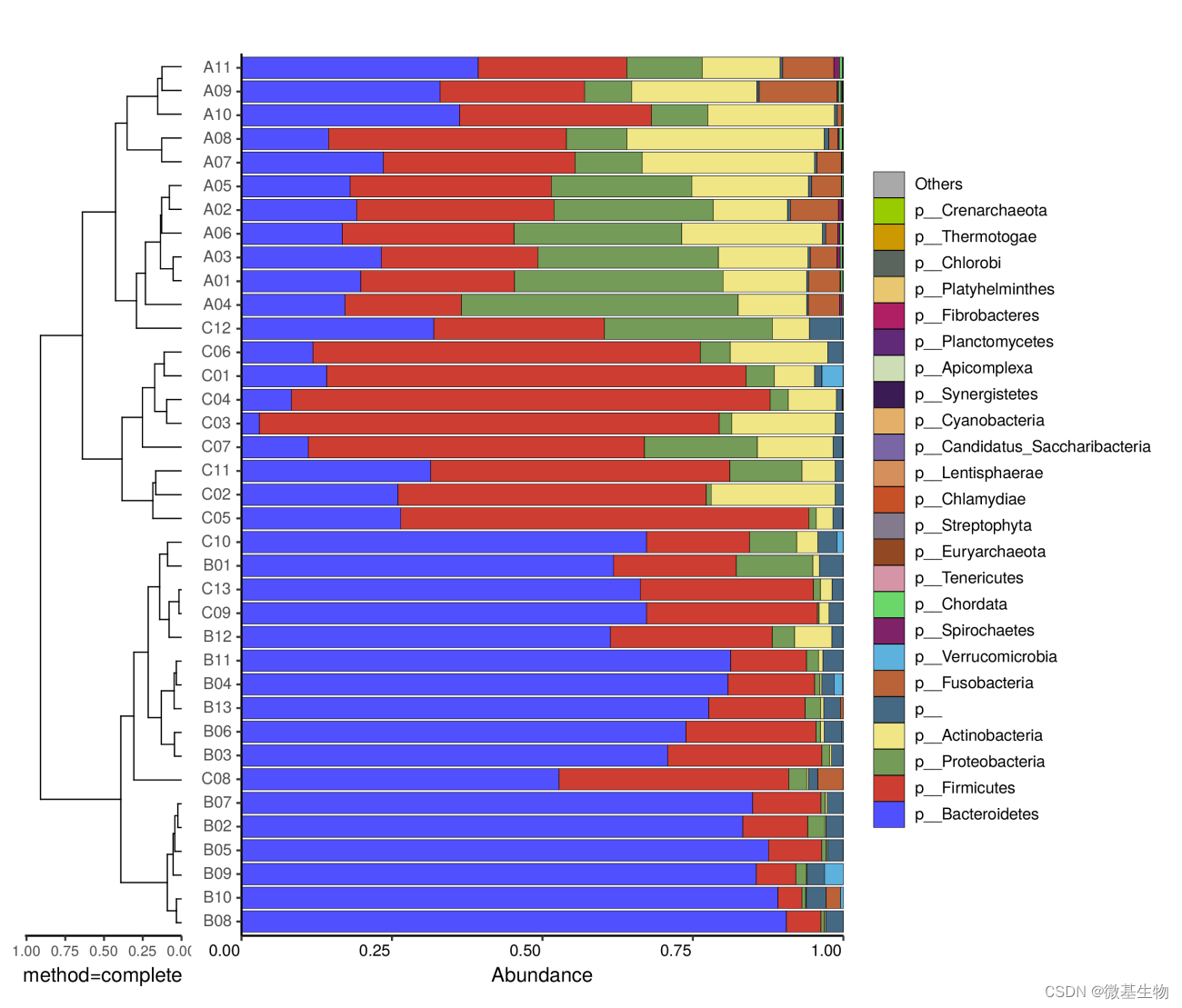
注:左边是样品间基于群落组成的层次聚类分析(Bray-curtis 算法),右边是样品的群落结构柱状图。
6差异分析
如果老师提供了分组信息,我们可以从物种、功能、alpha、beta多样性不同的角度进行差异分析。包括参数检验、非参数检验、lefse分析等。
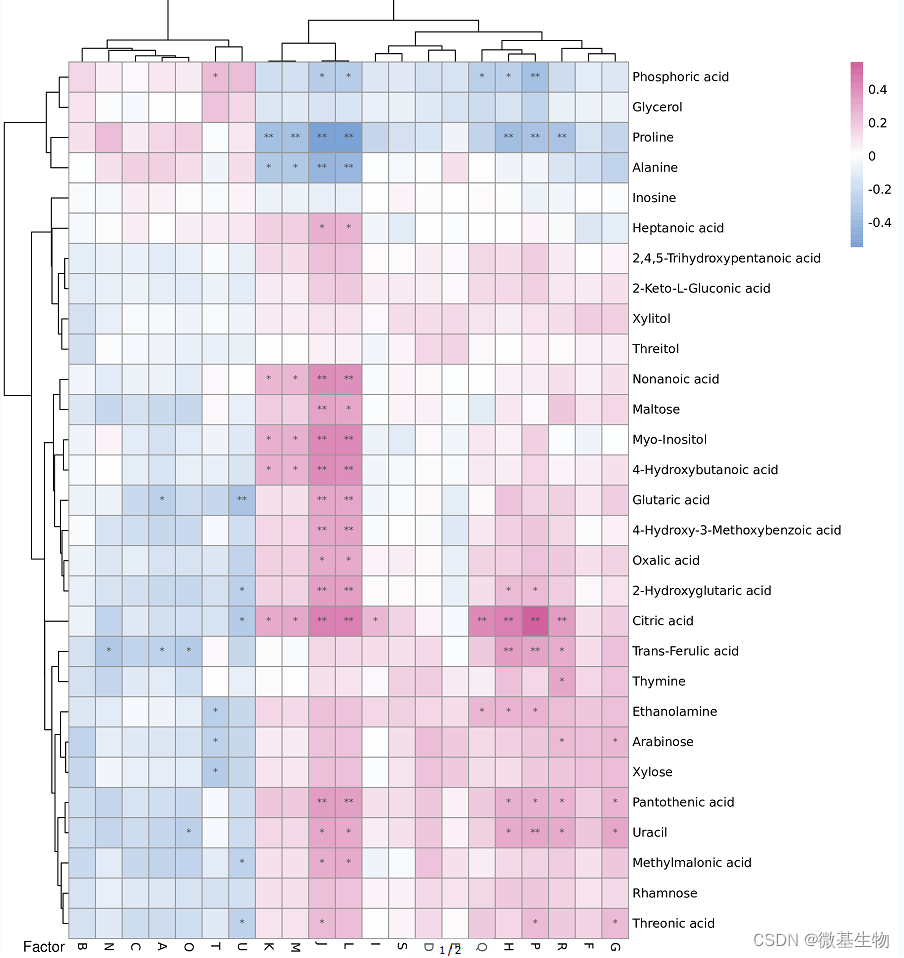
相关性热图
7 相关性分析
功能和物种之间的相关性、物种和临床指标之间的相关性,只要是同样的样本检测了两种不同的指标,都可以进行两个指标之间的关联分析。展示方式主要是spearman热图和网络图的形式。
图中
(1)颜色代表相关性系数:蓝色为负相关,且颜色越深,相关性系数越大,粉红色为正相关,且颜色越深,相关性系数越大,具体的颜色与相关性系数的对应关系见图中右上角的图注。
(2)横轴代表的是临床因子,纵轴代表的是代谢物。
(3)图中的*代表P值,*为0.05>P>0.01,**为0.01>P>0.001…..,只要是图中标注了*的,都是有显著相关的。
(4)左侧和上面的树都是根据相关性系数的相似性情况进行聚类的。
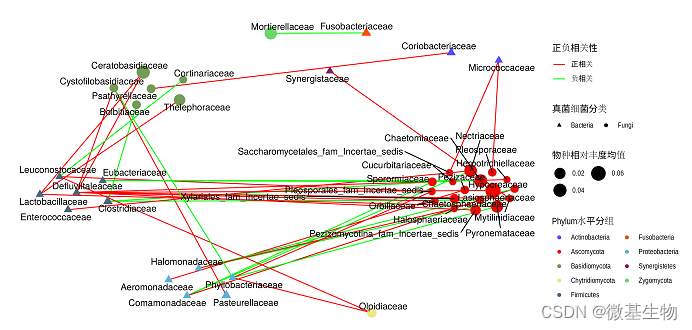
图中:
(1)图中形状代表不同的检测类型:本图是细菌和真菌,也可以是物种和不同功能之间的相关性。
(2)spearman的结果中p小于0.05,r绝对值大于0.6.
(3)节点的大小代表相对丰度的均值或是相关性程度。
(4)图中节点的颜色代表不同的门。
注意:网络图中展示的信息需要根据老师的要求来设计。