随着高通量技术的发展,宏基因组学(metagenomics)已经成为研究微生物群落物种及功能的前沿科学,在肠道微生物、环境微生物等研究领域具有广泛应用。宏基因组学通过对微生物群落全部DNA进行高通量测序,将测序序列与公共数据库进行比对或从头组装出微生物基因组,从而识别微生物群落的物种和功能基因。
目前主流的宏基因组数据分析方法包括三种:基于测序结果进行组装的分析方法;基于reads直接和已知数据库进行比对的方法;基于分箱bin的方法。
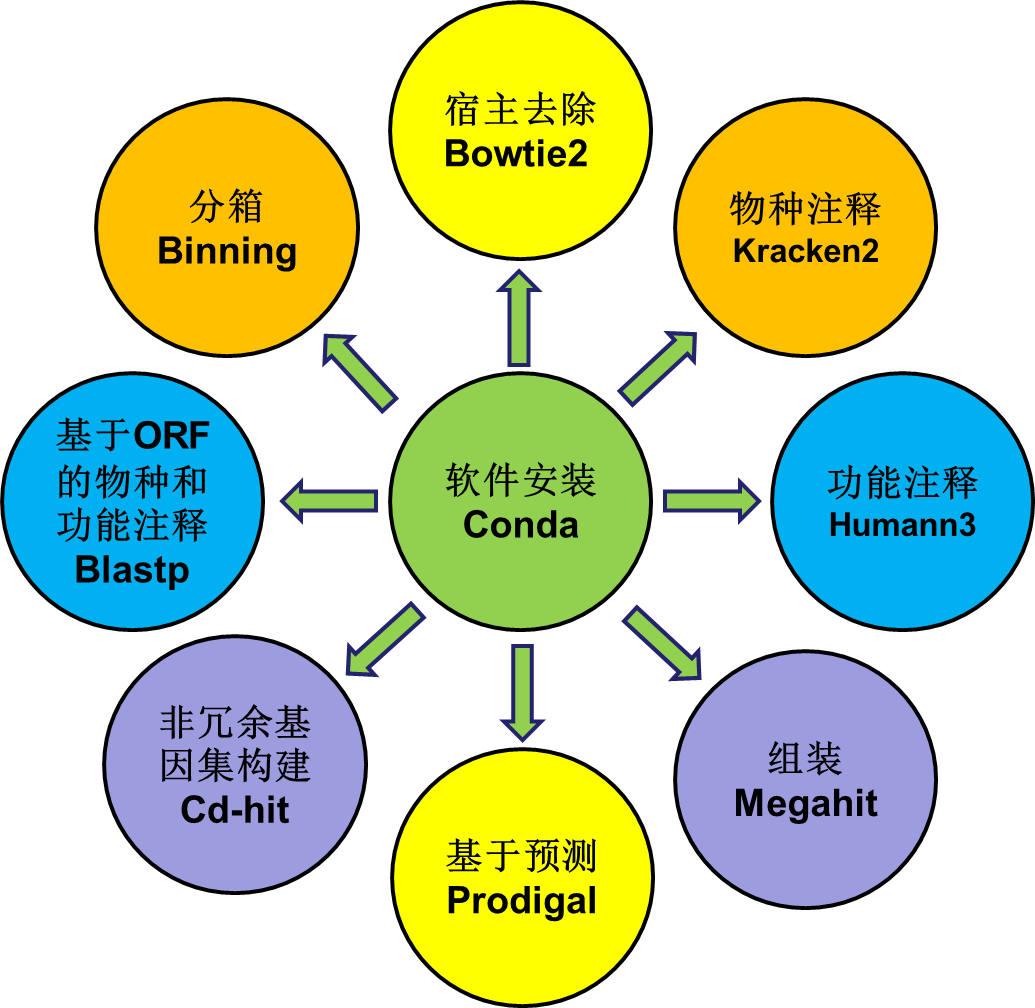
常用到的分析工具见下图:
一、基于组装
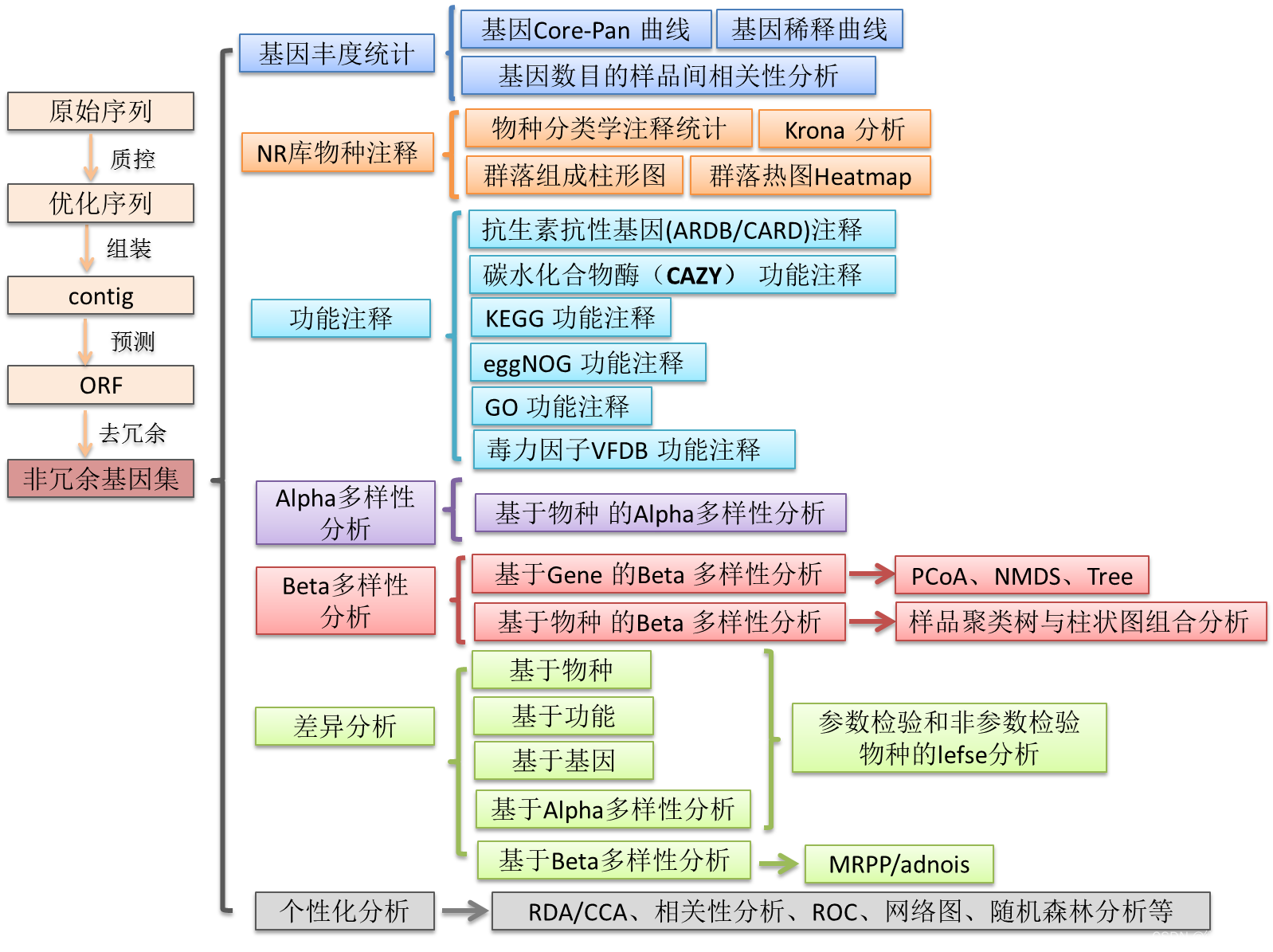
数据分析从下机原始序列开始,首先对原始序列进行去接头、 质量剪切以及去除污染等优化处理。然后使用优质序列进行拼接组装和基因预测,将各样本预测得到的基因集合并在一起去冗余,得到非冗余基因集;对得到的非冗余基因集与NCBI的nr数据库进行比对,得到物种信息,与各个功能数据库(抗性基因ARDB/CARD、碳水化合物CAZy数据库、KEGG数据库,eggnog数据库、VFDB数据库)进行比对,得到不同功能的注释结果。并使用BWA软件将优化序列比对到非冗余基因集,计算得到各基因在各样品中的丰度信息(RPKM); 最后对物种和功能注释结果进行统计及后继分析。
具体分析的流程见下图:
二、基于Reads
基于Reads的宏基因组分析,使用质控好的优化序列与已知物种和功能数据库进行比对,从而得到每个样本的物种、功能注释信息和相对丰度。此方法最大的优势是不用进行基因组的组装,从而快速的得到群落的物种与功能组成信息。
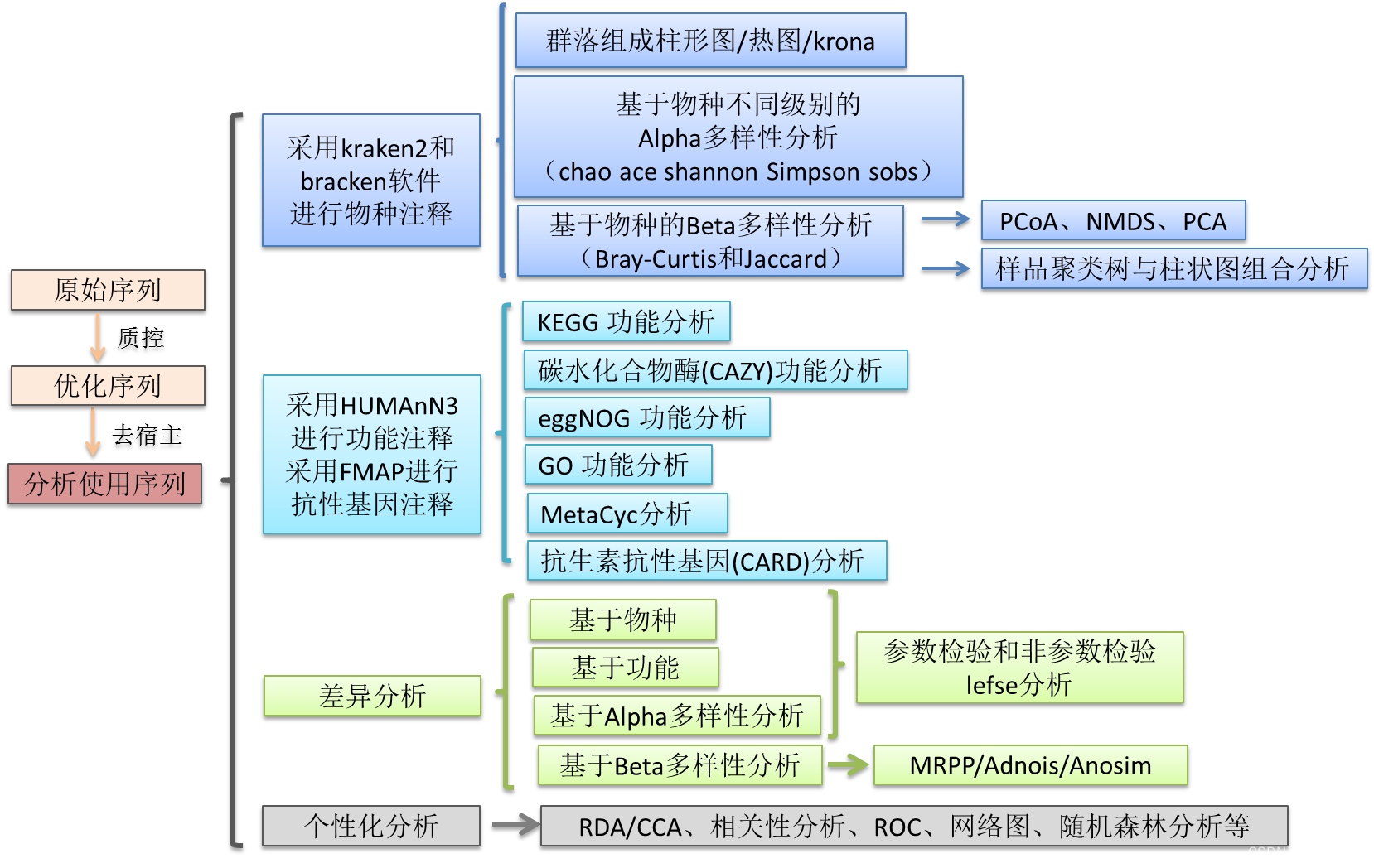
物种注释:从质控好的优化序列出发,使用Kranken2软件和微生物数据库(包括RefSeq最近的99版本基因组数据,涵盖细菌、真菌、病毒、古菌、原生动物)鉴别样本中所含的物种,再用Bracken对Kraken2得到的分类结果进性分类后贝叶斯重新估算丰度来估算宏基因组样本的种级别相对丰度;
功能注释:从质控好的优化序列出发,使用HUMAnN3软件和蛋⽩质数据库(UniRef90)进⾏⽐对,根据UniRef90 的ID 和各个功能数据库ID的对应关系,统计各个功能数据库对应功能相对丰度;
其他功能数据库:使⽤FMAP软件将各样本质控好的优化序列与参考数据库进⾏⽐对(基于DIAMOND),根据⽐对结果,统计出每个样本⽐对到各参考序列的reads数,从⽽计算相对丰度;
具体分析流程见下图:
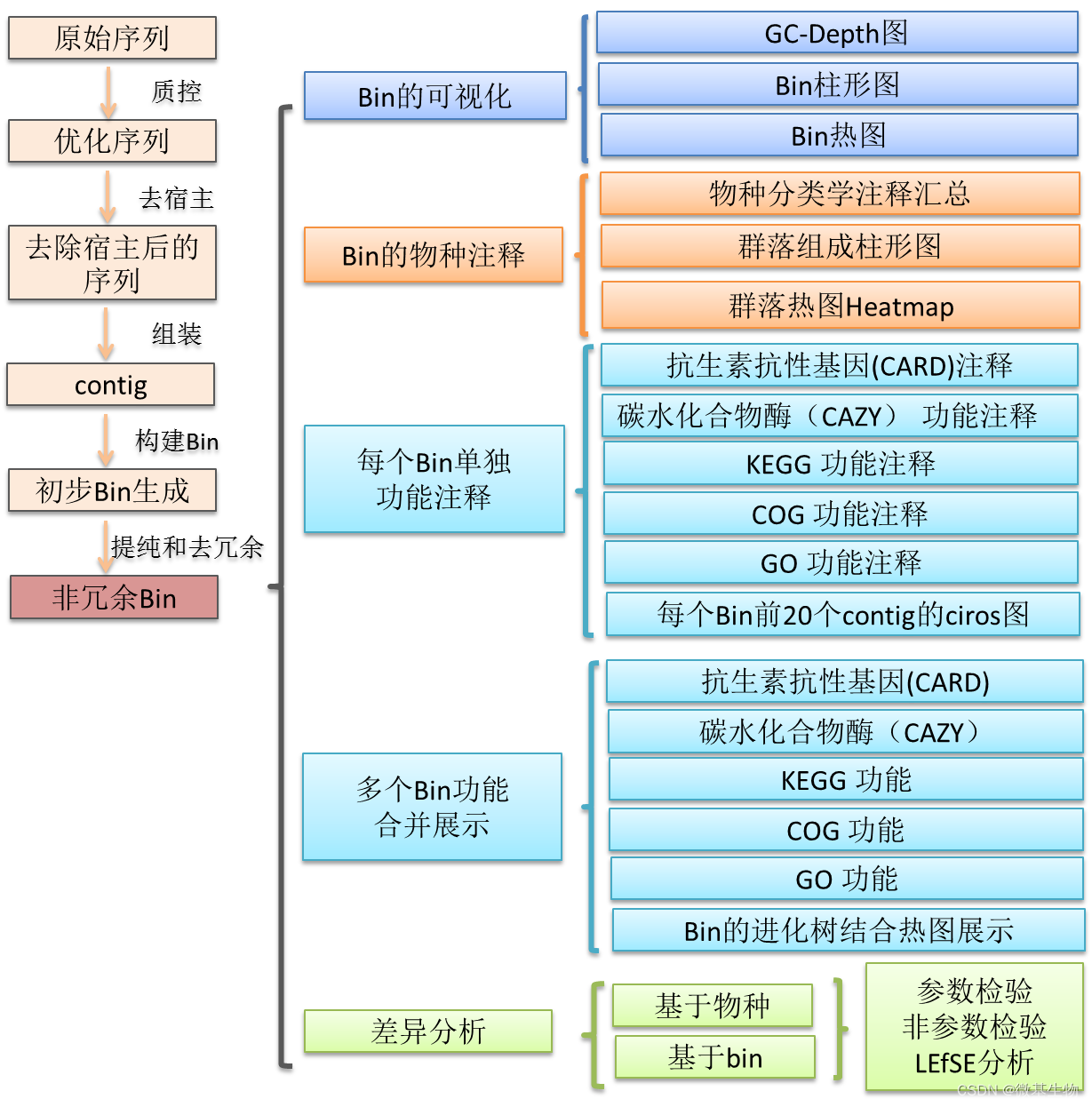
三、基于Bin分析
宏基因组分箱(Binning)是将序列组装得到的Contigs按物种分开归类的过程。基于Binning的宏基因组分析流程,数据分析从下机原始序列开始,首先对原始序列进行去接头、 质量剪切以及去除污染等优化处理。然后使用优质序列进行拼接组装得到Contigs;使用metabat2和maxbin2软件分别对每个样本的Contigs进行分箱;不同软件分箱得到的bin进行合并(binning_refiner)、提纯(MAGpurify);然后将所有样优化后的bin进行去冗余(dRep);而后分别从物种和功能方面进行信息统计。
具体分析流程见下图:
四、三种方法各自的优缺点:
1、基于组装的方法可以得到各个ORF的丰度信息,不管该ORF是否有对应的功能或是物种信息。如果是环境样本,微生物非常复杂,里面会得到的ORF信息量大,导致分析需要的时间延长。
2、基于Reads与已知序列Mapping的方法,没办法表征大量尚未分离和测序的微生物群体。现有的比对算法只能发现近缘关系,且仅能用于分析数据库中与基因组最密切相关的环境微生物。
但是基于基于Reads与已知序列Mapping的方法可以快速的得到物种和功能分析的结果。
3、Binning:Metagenome 组装完成后,我们得到的是成千上万的 contigs,我们需要知道哪些 contigs 来自哪一个基因组,或者都有哪些微生物的基因组。所以需要将 contigs 按照物种水平进行分组归类,称为 "bining"。主要依据是:来自同一菌株的序列,其核酸组成是相似的。优势:通过宏基因组测序尽可能完整的组装出样本中菌株的基因组。