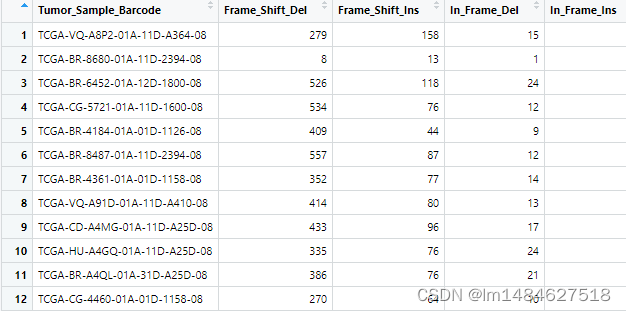
#1、准备文件/数据并加载相应的包
#1.1下载并加载相应的包,有就直接加载,没有就下载后再加载。
install.packages("pacman")
library(pacman)
p_load(TCGAbiolinks,DT,tidyverse)
BiocManager::install("TCGAbiolinks")
library(tidyverse)
BiocManager::install("maftools")
library(maftools)
library(dplyr)
#1.2需要准备亚型文件

#用之前用来构建风险模型的时候生成的具有高低风险组分组的文件
#行是样本名
#直接加载
load("F:/mypro/cancer-cuprosis/TCGA-STAD-cuprosis-lncrna/STAD-cuprosis-lncrna/mk-model/train_test_all_risk.RData")
View(all_surv_expr)
#就是上面截图这个样子。其实只需要有risk那一列就行了。
#1.3准备maf格式文件,并根据高低风险组将maf分为高低风险样本的亚型
raw<-GDCquery(
project= "TCGA-STAD",
data.category = "Simple Nucleotide Variation",
access = "open",
legacy = FALSE,
data.type= "Masked Somatic Mutation",
workflow.type = "Aliquot Ensemble Somatic Variant Merging and Masking"
)
#下载数据
GDCdownload(raw)
#获取数据
maf<- GDCprepare(raw)
#读取数据
maf<-maf %>% maftools::read.maf()
#一开始我以为把突变数据提取成表格就行了,后面发现不行。做瀑布图的时候,那个代码不能识别这样的表达矩阵,需要时maf格式的。
#找到样本所在的位置
maf@data$Tumor_Sample_Barcode
#改样本名
#样本名需要和亚型文件的样本名一致,好后面做提取处理。
mut$Tumor_Sample_Barcode<-substring(mut$Tumor_Sample_Barcode,1,12)
mut$Tumor_Sample_Barcode[1]
![]()
mut$Tumor_Sample_Barcode<-substring(mut$Tumor_Sample_Barcode,1,12)
mut.High <- mut[(mut$Tumor_Sample_Barcode %in% rownames(all_surv_expr)[all_surv_expr$risk=="high"]),]
### %in% 判断前面一个向量是否在后面一个向量中存在,返回布尔值
mut.Low <- mut[(mut$Tumor_Sample_Barcode %in% rownames(all_surv_expr)[all_surv_expr$risk=="low"]),]
maf.High <-read.maf(maf = mut.High,isTCGA = T)## 读取高风险亚型的突变数据
maf.Low <- read.maf(maf = mut.Low,isTCGA = T)## 读取低风险亚型的突变数据
maf.all <- read.maf(maf = mut,isTCGA = T)## 读取总的样本突变数据
# 下面设置颜色,人种等信息,这里的代码不需要修改:
col = RColorBrewer::brewer.pal(n = 10, name = 'Paired')
names(col) = c('Frame_Shift_Del','Missense_Mutation', 'Nonsense_Mutation', 'Frame_Shift_Ins','In_Frame_Ins', 'Splice_Site', 'In_Frame_Del','Nonstop_Mutation','Translation_Start_Site','Multi_Hit')
#人种
racecolors = RColorBrewer::brewer.pal(n = 4,name = 'Spectral')
names(racecolors) = c("ASIAN", "WHITE", "BLACK_OR_AFRICAN_AMERICAN", "AMERICAN_INDIAN_OR_ALASKA_NATIVE")
# 下面开始绘制总的瀑布图,代码及图片如下
oncoplot(maf = maf.all,
colors = col,#给突变配色
top = 20)
# 绘制高风险亚型瀑布图,代码及图片如下:
oncoplot(maf = maf.High,
colors = col,#给突变配色
top = 20)
# 绘制低风险瀑布图,代码及图片如下:
oncoplot(maf = maf.Low,
colors = col,#给突变配色
top = 20)