目录
- 简介
- 安装annotatr包
- 使用annotatr进行注释
- CpG注释
- 基因注释
- 自定义注释
- 读取基因组区域
- 注释区域
简介
下一代测序实验和生物信息学管道产生的基因组区域在注释基因组特征时更有意义。出现在外显子或增强子中的SNP可能比出现在基因间区域的SNP更令人感兴趣。有趣的是,我们发现一种特定的转录因子主要结合在启动子中,而另一种转录因子主要结合在3’非翻译区。含有CpG岛的启动子的超甲基化可能表明一种情况下与另一种情况下不同的调节机制。
annotatr提供了基因组注释和一组功能,用于在基因组注释的上下文中读取、相交、总结和可视化基因组区域。
安装annotatr包
使用Bioconductor可以安装annotatr,命令如下:
if (!requireNamespace("BiocManager", quietly=TRUE))install.packages("BiocManager")
BiocManager::install("annotatr")
可以通过GitHub存储库或Bioconductor获得annotatr的开发版本。用devtools包安装开发版本最简单,如下所示:
devtools::install_github('rcavalcante/annotatr')
详细的GitHub相关信息可以在GitHub releases找到描述。
使用annotatr进行注释
annotatr可以使用三种类型的注释:
1、内置注释包括CpG注释、基因注释、增强子、GENCODE lncRNAs和chromHMM中的染色质状态。以某种方式检索和处理这些注释中每一个的基本数据。有关数据源和处理的详细信息,请参见以下各项。
2、AnnotationHub注释包括Bioconductor AnnotationHub web资源中的任何GRanges资源。
3、用户提供的自定义注释。
CpG注释
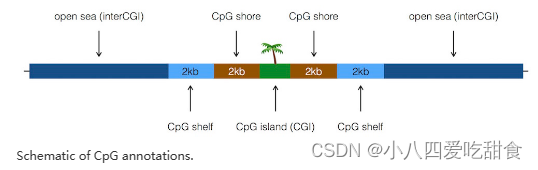
CpG岛是所有CpG注释的基础,由给定生物体的AnnotationHub包提供。CpG shores被定义为从CpG岛的末端开始的2Kb上游/下游,减去CpG岛。CpG shelves被定义为CpG shores的最远上游/下游限制的另一个2Kb上游/下游,减去CpG岛和CpG shores。剩余的基因组区域构成了CGI间注释。
shoresCpG注释可用于hg19、hg38、mm9、mm10、rn4、rn5和rn6。
基因注释
基因注释由来自GenomicFeatures的函数和来自TxDb和org.eg.db的数据确定。基因注释包括TSS上游1-5Kb、启动子(< TSS上游1Kb)、5’UTR、第一外显子、外显子、内含子、CDS、3’UTR和基因间区域(基因间区域不包括前面的注释列表)。下面的示意图说明了通过GenomicFeatures函数从TxDb包中提取的不同注释之间的关系。
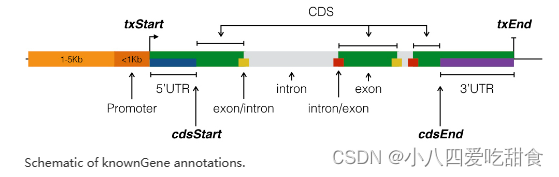
基因注释中还包括内含子和外显子内含子边界。这些注释位于外显子和内含子之间任何边界的上游/下游200bp处。值得注意的是,边界是相对于基因链而言的。
非基因间基因注释包括Entrez ID和基因符号信息(如果存在)。org*.eg.db包对于部分生物体可以提供基因id和基因符号。
基因注释已经填充了tx_id、gene_id和symbol列。它们分别是,已知基因转录物名称、Entrez基因ID和基因符号。
基因注释适用于所有hg19、hg38、mm9、mm10、rn4、rn5、rn6、dm3和dm6。
自定义注释
用户可以使用read_annotations()函数从BED文件中加载自己的注释,该函数使用rtracklayer::import()函数。输出是一个带有mcols()的GRanges,用于id、tx_id、gene_id、符号和类型。如果用户希望在自定义注释中包含tx_id、gene_id和/或symbol,则可以将它们作为额外的列包含在BED6输入文件中。
## Use ENCODE ChIP-seq peaks for EZH2 in GM12878
## These files contain chr, start, and end columns
ezh2_file = system.file('extdata', 'Gm12878_Ezh2_peak_annotations.txt.gz', package = 'annotatr')## Custom annotation objects are given names of the form genome_custom_name
read_annotations(con = ezh2_file, genome = 'hg19', name = 'ezh2', format = 'bed')print(annotatr_cache$get('hg19_custom_ezh2'))
## GRanges object with 2472 ranges and 5 metadata columns:
## seqnames ranges strand | id tx_id gene_id
## <Rle> <IRanges> <Rle> | <character> <logical> <logical>
## [1] chr1 860063-860382 * | ezh2:1 <NA> <NA>
## [2] chr1 934911-935230 * | ezh2:2 <NA> <NA>
## [3] chr1 3573321-3573640 * | ezh2:3 <NA> <NA>
## [4] chr1 6301401-6301720 * | ezh2:4 <NA> <NA>
## [5] chr1 6301996-6302315 * | ezh2:5 <NA> <NA>
## ... ... ... ... . ... ... ...
## [2468] chrX 99880950-99881269 * | ezh2:2468 <NA> <NA>
## [2469] chrX 108514101-108514420 * | ezh2:2469 <NA> <NA>
## [2470] chrX 111981673-111981992 * | ezh2:2470 <NA> <NA>
## [2471] chrX 118109216-118109535 * | ezh2:2471 <NA> <NA>
## [2472] chrX 136114771-136115090 * | ezh2:2472 <NA> <NA>
## symbol type
## <logical> <character>
## [1] <NA> hg19_custom_ezh2
## [2] <NA> hg19_custom_ezh2
## [3] <NA> hg19_custom_ezh2
## [4] <NA> hg19_custom_ezh2
## [5] <NA> hg19_custom_ezh2
## ... ... ...
## [2468] <NA> hg19_custom_ezh2
## [2469] <NA> hg19_custom_ezh2
## [2470] <NA> hg19_custom_ezh2
## [2471] <NA> hg19_custom_ezh2
## [2472] <NA> hg19_custom_ezh2
## -------
## seqinfo: 298 sequences (2 circular) from hg19 genome
要查看annotatr_cache环境中的内容,请执行以下操作:
print(annotatr_cache$list_env())
读取基因组区域
read_regions()使用rtracklayer::import()函数读入BED文件并将其转换为GRanges对象。普通BED文件中的name和score列可以分别用于分类数据和数值数据。此外,任意数量的分类和数字数据列可以附加到BED6文件中。extraCols参数用于此目的,rename_name和rename_score列允许用户为这些列提供更具描述性的名称。
# This file in inst/extdata represents regions tested for differential
# methylation between two conditions. Additionally, there are columns
# reporting the p-value on the test for differential meth., the
# meth. difference between the two groups, and the group meth. rates.
dm_file = system.file('extdata', 'IDH2mut_v_NBM_multi_data_chr9.txt.gz', package = 'annotatr')
extraCols = c(diff_meth = 'numeric', mu0 = 'numeric', mu1 = 'numeric')
dm_regions = read_regions(con = dm_file, genome = 'hg19', extraCols = extraCols, format = 'bed',rename_name = 'DM_status', rename_score = 'pval')
# Use less regions to speed things up
dm_regions = dm_regions[1:2000]
print(dm_regions)
## GRanges object with 2000 ranges and 5 metadata columns:
## seqnames ranges strand | DM_status pval diff_meth
## <Rle> <IRanges> <Rle> | <character> <numeric> <numeric>
## [1] chr9 10850-10948 * | none 0.5045502 -10.7329047
## [2] chr9 10950-11048 * | none 0.2227126 8.7195270
## [3] chr9 28950-29048 * | none 0.5530958 0.0700847
## [4] chr9 72850-72948 * | hyper 0.0116294 44.8753244
## [5] chr9 72950-73048 * | none 0.1752872 17.7606626
## ... ... ... ... . ... ... ...
## [1996] chr9 35605150-35605248 * | none 0.274255 -0.0539158
## [1997] chr9 35605250-35605348 * | none 0.918064 0.0329283
## [1998] chr9 35605350-35605448 * | none 0.614312 -0.0977500
## [1999] chr9 35605450-35605548 * | none 1.000000 0.0000000
## [2000] chr9 35605550-35605648 * | none 0.814567 0.0349967
## mu0 mu1
## <numeric> <numeric>
## [1] 79.981920 90.7148252
## [2] 86.704015 77.9844878
## [3] 0.124081 0.0539963
## [4] 72.455413 27.5800883
## [5] 28.440368 10.6797057
## ... ... ...
## [1996] 0.000000 0.0539158
## [1997] 0.328024 0.2950959
## [1998] 0.130184 0.2279345
## [1999] 0.000000 0.0000000
## [2000] 0.118272 0.0832756
## -------
## seqinfo: 298 sequences (2 circular) from hg19 genome
注释区域
用户可以通过builtin_annotations()快捷方式列出的访问器来选择注释,或者使用如上所述的定制注释。hg19_cpgs快捷方式将区域标注为CpG岛、CpG shores、CpG shelves和inter-CGI。hg19_basicgenes捷径将区域注释为1-5Kb、启动子、5’UTRs、外显子、内含子和3’UTRs。其他builtin_genomes()的快捷方式也以类似的方式访问。
annotate_regions()需要一个GRanges对象(read_regions()的结果或现有对象)、一个批注的GRanges对象和一个逻辑值,该值指示在调用GenomicRanges::findOverlaps()时是否忽略. strand。正整数minoverlap也被传递给GenomicRanges::findOverlaps()并指定要分配给注释的区域所需的最小重叠。
在注释区域之前,必须用build_annotations()构建它们,这需要一个所需注释代码的字符向量。
# Select annotations for intersection with regions
# Note inclusion of custom annotation, and use of shortcuts
annots = c('hg19_cpgs', 'hg19_basicgenes', 'hg19_genes_intergenic','hg19_genes_intronexonboundaries','hg19_custom_ezh2', 'hg19_H3K4me3_Gm12878')# Build the annotations (a single GRanges object)
annotations = build_annotations(genome = 'hg19', annotations = annots)# Intersect the regions we read in with the annotations
dm_annotated = annotate_regions(regions = dm_regions,annotations = annotations,ignore.strand = TRUE,quiet = FALSE)
# A GRanges object is returned
print(dm_annotated)
## GRanges object with 11374 ranges and 6 metadata columns:
## seqnames ranges strand | DM_status pval diff_meth
## <Rle> <IRanges> <Rle> | <character> <numeric> <numeric>
## [1] chr9 10850-10948 * | none 0.504550 -10.73290
## [2] chr9 10850-10948 * | none 0.504550 -10.73290
## [3] chr9 10950-11048 * | none 0.222713 8.71953
## [4] chr9 10950-11048 * | none 0.222713 8.71953
## [5] chr9 10950-11048 * | none 0.222713 8.71953
## ... ... ... ... . ... ... ...
## [11370] chr9 35605550-35605648 * | none 0.814567 0.0349967
## [11371] chr9 35605550-35605648 * | none 0.814567 0.0349967
## [11372] chr9 35605550-35605648 * | none 0.814567 0.0349967
## [11373] chr9 35605550-35605648 * | none 0.814567 0.0349967
## [11374] chr9 35605550-35605648 * | none 0.814567 0.0349967
## mu0 mu1 annot
## <numeric> <numeric> <GRanges>
## [1] 79.9819 90.7148 chr9:6987-10986:+
## [2] 79.9819 90.7148 chr9:1-24849:*
## [3] 86.7040 77.9845 chr9:10987-11986:+
## [4] 86.7040 77.9845 chr9:6987-10986:+
## [5] 86.7040 77.9845 chr9:1-24849:*
## ... ... ... ...
## [11370] 0.118272 0.0832756 chr9:35605281-35605616:+
## [11371] 0.118272 0.0832756 chr9:35605281-35605835:+
## [11372] 0.118272 0.0832756 chr9:35605281-35605835:+
## [11373] 0.118272 0.0832756 chr9:35605281-35605835:+
## [11374] 0.118272 0.0832756 chr9:35603969-35605991:*
## -------
## seqinfo: 298 sequences (2 circular) from hg19 genome
annotate_regions()函数返回一个GRanges,但是操作一个强制的data.frame可能更方便。
# Coerce to a data.frame
df_dm_annotated = data.frame(dm_annotated)# See the GRanges column of dm_annotaed expanded
print(head(df_dm_annotated))
## seqnames start end width strand DM_status pval diff_meth mu0
## 1 chr9 10850 10948 99 * none 0.5045502 -10.73290471 79.981920
## 2 chr9 10850 10948 99 * none 0.5045502 -10.73290471 79.981920
## 3 chr9 10950 11048 99 * none 0.2227126 8.71952705 86.704015
## 4 chr9 10950 11048 99 * none 0.2227126 8.71952705 86.704015
## 5 chr9 10950 11048 99 * none 0.2227126 8.71952705 86.704015
## 6 chr9 28950 29048 99 * none 0.5530958 0.07008468 0.124081
## mu1 annot.seqnames annot.start annot.end annot.width annot.strand
## 1 90.7148252 chr9 6987 10986 4000 +
## 2 90.7148252 chr9 1 24849 24849 *
## 3 77.9844878 chr9 10987 11986 1000 +
## 4 77.9844878 chr9 6987 10986 4000 +
## 5 77.9844878 chr9 1 24849 24849 *
## 6 0.0539963 chr9 28923 29077 155 *
## annot.id annot.tx_id annot.gene_id annot.symbol
## 1 1to5kb:34327 uc011llp.1 100287596 DDX11L5
## 2 inter:8599 <NA> <NA> <NA>
## 3 promoter:34327 uc011llp.1 100287596 DDX11L5
## 4 1to5kb:34327 uc011llp.1 100287596 DDX11L5
## 5 inter:8599 <NA> <NA> <NA>
## 6 H3K4me3_Gm12878:27530 <NA> <NA> <NA>
## annot.type
## 1 hg19_genes_1to5kb
## 2 hg19_cpg_inter
## 3 hg19_genes_promoters
## 4 hg19_genes_1to5kb
## 5 hg19_cpg_inter
## 6 hg19_H3K4me3_Gm12878
以上是用到的部分,只需准备bed文件(chr,start,end三列)就可以获得各种信息。
参考链接:http://mirrors.nju.edu.cn/bioconductor/packages/3.13/bioc/vignettes/annotatr/inst/doc/annotatr-vignette.html#reading-genomic-regions