Protein & Cell综述:基于R语言的微生物组数据挖掘的最佳流程
近日,中国农业科学院刘永鑫组联合南京农业大学袁军组在国际期刊 Protein & Cell (IF = 15.3) 发表了题为”“The best practice for microbiome analysis using R”的综述论文,建立了微生物组分析的R语言代码库EasyMicrobiomeR,助力微生物组学发展。

IF: 15.328
DOI: https://doi.org/10.1093/procel/pwad024
上线时间: 2023/05/02
第一作者:
文涛(Tao Wen),牛国庆(Guoqing Niu)
通讯作者:
袁军Jun Yuan)(junyuan@njau.edu.cn)、刘永鑫(Yong-Xin Liu)(liuyongxin@caas.cn)
合作作者:
陈同( Tong Chen),沈其荣(Qirong Shen)(shenqirong@njau.edu.cn)

大众评审
同行评审通常只有2-4个审稿人,样本量不足可能会遗漏一些问题。我们开展大众评审模式,对同行评审的文章进行补充,希望进一步提高文章质量。
文章目前处于提前在线发表阶段,正式文档正在排版校对中。欢迎广大读者下载文章Proof: http://www.imeta.science/iMeta/temp/PROCEL_pwad024_wt.pdf ,提交修改建议至 【金山文档】 文章大众评审
https://kdocs.cn/l/cnUufkyt5F5A 中帮忙文章质量提高。
对于较重要的修改意见,将会在文章中致谢作者姓名。有兴趣对本项目新增功能和贡献代码的用户长期招募、欢迎加入,共同参与EasyMicrobiomeR下一版开发和共同发表,共同打造本领域基础设施,推动微生物组学发展。
图文摘要
本文详细介绍了基于R语言的324个常用R包进行微生物组数据挖掘的过程。特别关注了六个微生物组分析集成R包(phyloseq, microbiome, MicrobiomeAnalystR, Animalcules, microeco, amplicon)。并提出了基于R语言进行微生物组分析的最佳流程,相关代码都可以在 https://github.com/taowenmicro/EasyMicrobiomeR 获取。如果觉得项目有用,欢迎点击GitHub主页右上角Star支持本项目。
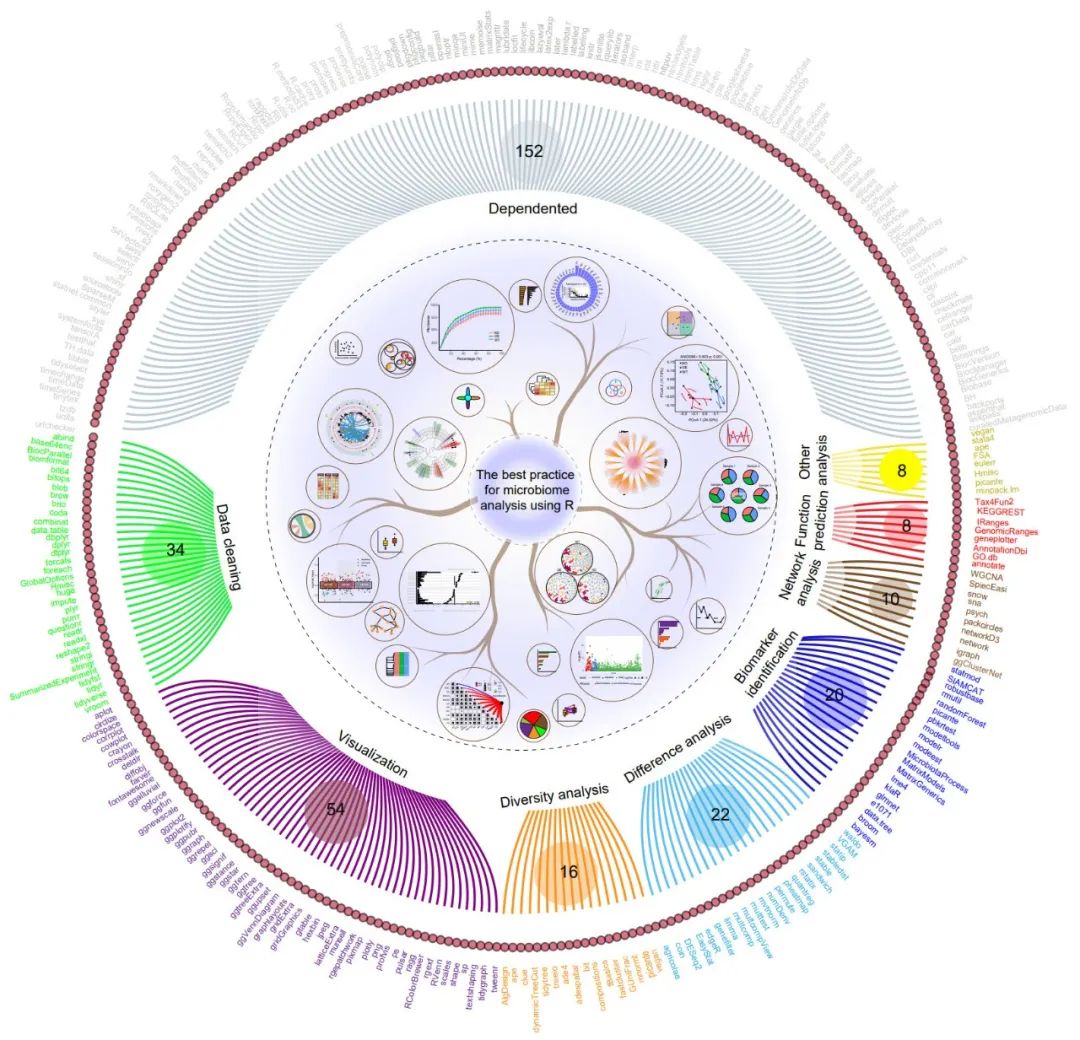
摘要
随着测序技术的逐步成熟,许多微生物组研究成果相继发表,推动了相关分析工具的出现和发展。R语言是目前广泛使用的用于微生物数据分析的平台,具有强大的功能。然而,数以万计的R包和无数类似的分析工具给许多研究人员挖掘微生物组数据带来了重大挑战。如何从众多的R包中选择合适、高效、方便、易学的工具已经成为许多微生物组研究人员面临的问题。我们整理了324个用于微生物组分析的常用R包,并根据应用类别(多样性、差异性、生物标志物、相关性和网络、功能预测等)对其进行分类,以帮助研究人员快速找到用于微生物组分析的相关R包。此外,我们对微生物组分析的集成R包(Phyloseq、Microbiome、MicrobiomeAnalystR、Animalcules、Microeco和Amplicon)进行了系统的分类,并总结了它们的优点和局限性,以帮助研究人员选择合适的工具。最后,我们对用于微生物组分析的R包进行了全面的回顾,总结了微生物组中大多数常见的分析内容,构建最适合微生物组分析的流程。本文附带了GitHub中数百个代码的例子,可以帮助初学者学习,也可以帮助分析人员比较和测试不同的工具。本文对R在微生物组中的应用进行了系统的梳理,为今后开发更好的微生物组工具提供了重要的理论依据和实践参考。所有代码都可以在GiHub:github.com/taowenmicro/EasyMicrobiomeR 上找到。
关键词: R包,微生物组,数据分析,可视化,扩增子,宏基因组
文章亮点
本文按照六大功能类别(多样性、差异性、生物标志物、相关性和网络、功能预测和其他分析)对324个常用R包进行功能分类;
对微生物组分析的集成R包(phyloseq, microbiome, MicrobiomeAnalystR, Animalcules, microeco, and amplicon)进行了系统的介绍,并总结了其优点和局限性;
本文总结了微生物组数据分析中常见的内容,并提供了一套最适合微生物组数据挖掘的分析流程;
在GitHub上,分享了包含上万行代码的例子,这些例子不仅可以帮助初学者学习,还可以帮助专业人员比较和测试不同的工具。
引言
杨盛蝶翻译,南京农业大学,博士在读;
宏基因组分析通过对微生物群落的DNA或RNA序列进行测序、定量、注释和分析,来研究微生物的多样性、结构和功能。微生物组研究中常用的高通量测序技术主要有扩增子测序(amplicon sequencing)和随机宏基因组测序(shotgun metagenomic sequencing)。扩增子测序以其成本低、分析体系成熟、分析过程简单等优点被广泛应用于微生物组研究中。随机宏基因组测序提供了微生物的功能信息和更准确的微生物组成信息,测序成本较高,所需计算资源较大。我们在之前的综述中系统地总结了这两种测序的详细流程(刘永鑫等,2021年,Protein & Cell)。微生物群落作为生物多样性的重要组成部分,在生物学、生态学、生物技术、农业和医学等领域发挥着至关重要的作用。微生物群落分析需要多种生物信息学方法,主要包括三个部分:1)数据预处理,2)量化和注释,3)统计和可视化(图1A)。在预处理阶段,对原始数据进行过滤和质量控制,以确保数据质量。在量化和注释步骤中,使用工具和数据库来识别微生物代表序列并注释微生物分类和功能。微生物群落分析的前两部分已经进行了很好的讨论,根据我们之前的论文(刘永鑫等,2023,iMeta)可以很好地完成这两部分。最后,在统计和可视化步骤中,使用各种统计方法来探索微生物群落的多样性、结构和潜在功能。
图1. 微生物群落数据分析工作流程及相关R软件包。
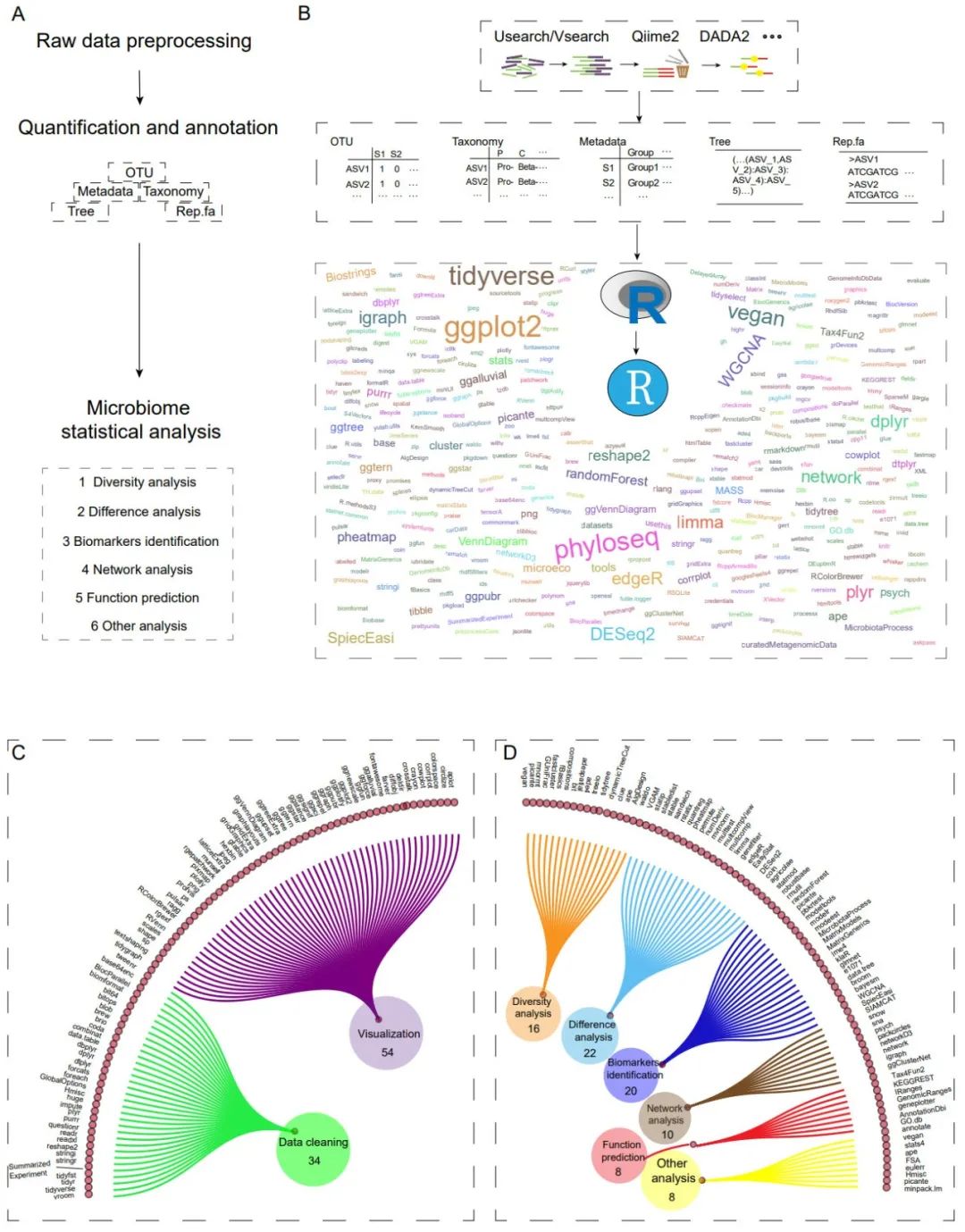
(A) 微生物群落数据分析工作流程概述。核心文件是特征表(OTU)、分类表、样品元数据(Metadata)、系统发育树(Tree)和代表序列(Rep.fa)。(B) 详细的微生物群落分析工作流程。首先,原始数据可以通过使用USEARCH/VSEARCH、QIIME2、DADA2软件包进行处理。然后,将重要文件保存,用于RStudio软件下的R语言环境开展下游分析。许多微生物分析方法都依赖于R语言开发的R包。词云图中的字体大小代表R包的引用次数。(C) 用于数据清理/处理和可视化的常用的R包。(D) 微生物群落分析R包划分为六个类别。
随着高通量测序技术的发展,许多研究利用扩增子技术(Thompson等,2017;Proctor等,2019)和随机宏基因组测序技术(Carrión等,2019;Li等,2022;Paoli等,2022年),这导致了微生物组分析方法学、软件和流程的开发,例如QIIME(Caporaso等,2010年)、Mothur(Schlos等,2009年)、USEARCH(Edgar,2010)、VSEARCH (Rognes等,2016)、QIIME 2(Bolyen等,2019年)、Parallel‐Meta Suite (Chen等,2022)、EasyAmplicon(Liu等,2023)、Kraken(Wood和Salzberg等,2014)、MEGAN (Huson等,2007)、MetaPhlAn2(Truong等,2015),HUMAnN2 (Franzosa et al.,2018)等。作为扩增测序数据分析最关键和最基本的步骤,OTU(操作分类单元)聚类方法在2015年前流行起来,而非聚类方法则是在最近几年逐步发展和广泛应用的。目前,常见的非聚类方法包括DADA2(Callahan等,2016)、deblur(Amir等,2017)、unoise3(Edgar和FlyvbJerg,2015)。其中最具代表性的非聚类算法之一是DADA2,它是用R语言创建的。这使得R语言(Ihaka和Gentleman,1996)在扩增子测序的原始数据处理中占据了重要地位。与许多可用于微生物群落测序数据分析的上游步骤的软件相比,下游的分析步骤严重依赖于R语言和各种软件包。这些分析主要包括:1)多样性分析;2)差异分析;3)相关性和网络分析;4)生物标志物识别;5)功能预测;6)微生物群落与其他指标的整合分析(包括系统发育分析和环境因素分析等)。除了多元变量统计分析之外,R中还有各种数据清洗包允许在不同的分析之间转换数据。
R是用于数据统计分析和可视化的自由、开源语言和环境,由新西兰奥克兰大学的Ross Ihaka和Robert Gentleman创建,现在由“R Development Core Team”负责。与SPSS、MINITAB, MATLAB等更适合处理标准化数据的分析工具相比,R语言既能处理已经被修改的数据,也能处理原始数据。R可以很容易地实现几乎所有的分析方法,许多最新的方法或算法都是最先在R上展示的。此外,R显示了出色的数据可视化,特别是对于复杂数据。强大而灵活的交互分析也是R的一个优势,同时支持可视化的数据探索。R语言的功能在很大程度上依赖于数以千计的R包,它们提供了各种各样的数据处理和分析的策略,几乎允许在R中完成任何数据分析过程。CRAN上发布的R包总数为18,981个,Bioconductor上发布的R包总数为2,183个(截至2023年1月31日)。这些程序包展示了R强大的数据处理和分析性能。
近年来,在R平台上开发了大量用于微生物组下游分析的R包,为相关研究领域做出了重要贡献。然而,下游分析R包的数量已经达到了令人眼花缭乱的水平(图1B)。此外,包含大量微生物组分析的集成R包逐渐出现,如phyloseq(McMurdie和Holmes,2013)、microeco(Liu等,2020)和amplicon(Liu等,2023)。如此丰富的R包为微生物组分析人员提供了更多的选择,但也使得在许多类似的分析工具中确定最合适的工具存在难度。此外,过多的R包使初学者很难走上一条组织良好的微生物组分析学习之路。因此,当务之急是比较相似的分析功能,并提取其异同点,选择最适合微生物组分析的流程,帮助初学者更有效地学习。
本文试图对324个常见的R包(附图1)进行分类和运行。特别是用于微生物组分析的集成R包,并完成以下三个部分:1)根据微生物组分析的功能类别比较不同的R包分析流程,分析结果并总结范例代码;2)根据微生物组分析的功能类别介绍六个集成R包的内容,比较分析结果,生成范例代码;3)根据所有R包,使用R语言选择最优的分析方法,并提供范例代码供研究人员参考和学习。
微生物数据分析前准备工作
微生物组下游分析需要准备五个数据文件,包括特征表、特征注释文件、样本分组文件、系统发育树和代表性序列。对于初学者来说,重要的是了解这些文件的格式和基本数据结构,并学习如何将这些文件导入R语言。此外,不同的分析内容往往对数据有不同的要求,需要学习一些数据操作技巧来满足各种功能的需求。最后,有必要学习R出图的基础知识,以便于展示结果。
数据准备和清洗
在对序列进行预处理、量化和注释后,需要对输出的文件进行进一步的分析,包括导入这些文件、清理数据、转换格式,这是后续R中微生物组分析所需的。在进行统计分析之前,必须掌握R语言的基本程序,以应对不同R包的数据输入要求。这一部分包括:数据的导入、整理、过滤、基本计算、转换、均一化和修改。原始数据处理中经常使用五种数据形式,包括特征表(文件格式为.csv/.txt/.xlsx/.biom,通常为分类和功能表,包括OTU/ASV/分类/基因/模块/路径表)、特征注释(.csv/.txt/.xlsx/.biom)、样本分组文件(.csv/.txt)、进化/系统发育树(.nwk/.tree)、代表性序列(.fast a/.fas/.fa)。所有与数据清理相关的包如图1C所示。微生物群落的表格数据输入主要是使用utils包(代码1A,GitHub中的脚本)中的read.table()、read.delim()和read.csv()等函数完成的。进化树文件的读取依赖于ape/ggtree/treeio包中的read.tree()或Phyloseq包中的read_tree()之类的函数。为了读取微生物组中具有代表性的序列文件,通常使用Biostrings包(Pages等,2016)中的readDNAStringSet()。目前,微生物组的大数据集成已成为一种趋势,并导致出现了用于整合多项研究数据的R包,如curatedMetagenomicData(Pasolli等,2017年)。只需要导入该R包,就可以重新分析筛选的数据,而不是输入的原始数据。
数据整理的本质可以概括为三个步骤:数据分割、使用函数处理以及将输出结果合并为所需的格式。R中基础包的函数组合在一起,可以满足微生物组数据操作的大部分要求。例如,结合基本统计函数[sum()、mean()、sd()等]的“for循环”,可用于进行微生物相对丰度(代码1B)的基本统计分析和数据转换;base包提供apply系列函数,包括apply()、sapply()、lapply()、Tapply()、aggregate()等,可以快速完成数据处理的三个阶段。apply家族函数提供了一个框架,起到替代for循环的作用,比R(代码1B)中的基本“for loop”函数快得多。类似的purr(https://github.com/tidyverse/purrr))包也可以代替“for loop”来执行高效的操作。
plyr包在base包的基础上升级了数据框,列表等多种数据形式的数据处理过程。plyr包提供了在一个函数内同时完成“Split - Apply - Combine”三个数据处理阶段,并且,plyr包实现R类型(向量、列表和数据框)之间的分组变换,基本上可以取代base包中的apply家族函数。可以很方便的处理分组计算,例如,计算不同分类水平下的微生物丰度(示例二)。reshape2包在数据处理过程中提供了长宽格式转化。由于ggplot2包大多数建模函数如lm()、glm()、gam()等会使用的是长数据格式,而微生物组数据一般都是宽数据格式,所以使用reshape2可以完成微生物组数据到绘图的转化。
dplyr包是tidyverse家族的一员,创新性地放弃了R中数据保存的常见形式,使用tibble格式(比data.Frame格式更强大)进行数据处理,可以更高效地完成行和列内的数据框选择、合并和统计,以及数据框长度和宽度格式的变化,配合%>%管道符号可以完成更复杂的数据处理过程。Tibble格式可以在分析和建模过程中存储数据,这对数据分析非常重要。例如,我们演示了使用dplyr和流程来运行随机森林建模以及重要变量的挑选过程(代码1E)。
R语言中的可视化
牛国庆翻译,南京农业大学,博士在读;
在大多数情况下,微生物组数据分析中,我们通常习惯于绘制标准图形,例如alpha/beta多样性和分类组成。图1C中显示是与可视化相关的R包。由于ggplot2的广泛使用(代码2A),出现了许多基于ggplot2扩展的R包,具有丰富的绘图样式、颜色和主题。这些R包主要包括用于绘制三元图(Hamilton和Ferry,2018)的ggtern(代码2B)、用于绘制网络图(Si等,2022)的ggraph(代码2C)、用于绘制进化树或物种分类图(Xu等,2022)的ggtree(代码2D)、ggalluvial包、ggVennDiagram包(代码2E),用于绘制饼图ggstatsplot包以及提供各种不同主题和颜色的输出的ggpubr包。此外,基于grid绘图系统的pheatmap和ComplexHeatmap软件包(GU,2022)可以绘制不同样本中特征的相对丰度(代码2F),VennDiagram包(Chen和Boutros,2011)可以显示不同样本中特征的数量。UpSetR包(Conway等,2017)可以绘制了一种类似于Venn图的新型图形,称为Upset视图。基于base的绘图系统虽然比较复杂并且难于学习,但是功能却非常强大,对于绘制复杂图形(如由微生物组成的和弦图),是一个不错的选择,例如使用circlize(Gu等,2014)包(代码2G)绘制微生物组成的弦图。
此外,经常有许多涉及图形组合的微生物组绘图工作。目前,R中有许多可以组合图形的工具,如cowplot、patchwork和aplot。patchwork包具有最强大的功能,支持模块化拼接图形(代码2H)。
微生物群落分析
我们在图1D中将微生物组数据分析分为以下六个主要类型:多样性分析、差异分析、生物标志物鉴定、相关性和网络分析、功能预测以及其他微生物组分析(包括朔源分析、群落组装过程以及微生物群落与环境因素之间的关联分析)。然后,我们将会对所有相关的R包进行组织、比较和总结。
多样性分析
微生物群落多样性主要包括Alpha多样性(Richness、Shannon、Simpson、Chao1、ACE等)、稀释曲线、Beta多样性(排序和聚类分析)、分类或功能组成。这里必须介绍一下vegan包(Oksanen等,2007),它是由包括来自芬兰的Oksanen在内的九位数量生态学家编写的,是一款最初用于处理群落生态学数据的工具。该包提供了各种数据标准化和转换方法。例如,用于Alpha多样性分析的数据可以使用rrarefy()函数在相同测序深度下进行标准化,用于排序分析的数据可以使用decostand()函数进行标准化处理(代码3A)。测序数据经过样本归一化后,多样性计算结果会更合理。此外,还可以使用adespatial(Dray等,2018)、ade4(Dray和Dufour,2007)和picante包(Kembel等,2010)来计算Alpha多样性指标。例如,可以使用picante包中的pd()函数计算系统发育多样性(代码3A)。vegan不仅可以进行Alpha多样性分析,还提供了一些其他功能,例如rda()函数用于主成分分析(PCA)和冗余分析(RDA),cca()函数用于对应分析(CA)和典型对应分析(CCA),decorana()函数用于决策曲线分析(DCA),metaMDS()函数用于行微生物组排序分析的非度量多维尺度分析(NMDS)(代码3B)。stats包中的prcomp()函数可用于主成分分析(PCA),这是一种降维分析方法。MASS包中提供的mca()函数和FactoMineR包中提供的MCA()函数可用于多重对应分析(代码3B);ape包提供的pcoa()函数可用于主坐标分析(PCoA);MASS包提供的lda()函数可用于线性判别分析(LDA,代码3C)。在运行多个排序操作之前,通常需要进行群落聚类。vegan包中的vegdist()函数可计算euclidean,manhattan, bray, canberra等多种生态距离(代码3B)。此外,距离计算也可以使用stats包中的dist()函数完成。除了排序分析,距离矩阵还可用于聚类分析。stats包中的hclust()函数可用于聚类分析,factoextra包、kmeans包也可以实现类似的功能(代码3D)。微生物组成分析主要用于显示微生物的丰度,需要使用dplyr包对数据进行整理,然后使用ggplot2包进行展示。
差异分析
差异分析分为群落水平分析和特征水平(包括任何分类和功能层次)分析。群落水平的差异分析主要使用函数包括:vegan包中的adonis()、anosim()、mrpp()函数以及ape包中的mantel.test()函数(代码4A)。特征水平的差异分析中组成差异可以利用stats包中的wilcox.test()函数(代码4B)和t.test()函数(代码4C)。随后,针对测序数据专门开发了数据矫正算法,例如edgeR包中的:Upper Quartile(UQ),Trimmed Mean of M-values (TMM),Relative Log Expression (RLE);DESeq2包中的Median(MED)以及metagenomeSeq(https://github.com/sirusb/metagenomeSeq)
包中Cumulative-Sum Scaling (CSS)算法(代码4F)。此外,ALDEx2包提供了多项式模型,可用于推断特征丰度并使用非参数检验、t检验或广义线性模型计算特征差异(代码4G)。ANCOM-BC包试图通过对数线性模型矫正偏差来解决样本异质性问题。此外,用于微生物组数据矫正和差异检验的其他R包括limma(代码4H)、DR、ANCOM(Lin和Peddada,2020)(代码4I)、corncob(代码4J)、Maaslin2(代码4K)等。Nearing等(2022)表明,他们比较了这些差异分析方法,并提出ALDEx2和ANCOM-II(anchom_v2.1.R,代码4L)是微生物群落差异分析中最好的方法。至于显著性检验,不同的包使用不同的方法进行显著性检验。例如,edgeR包使用Fisher检验,DESeq2和corncob包使用Wald检验,limma包使用t检验。还有其他的显著性检验方法,如Wilcoxon秩和检验(ALDEx2和ANCOM-II),方差分析(ANOVA)(Maaslin2)等。
生物标志物判断
特征微生物的寻找是为了解释某些问题,如肥胖或高血压人群的肠道生物标志物,或镰刀菌枯萎病发病土壤的生物标志物等。通过差异分析挑选的微生物通常无法确定它们是否是关注的主要差异。因此,权重分析或机器学习方法被用来进一步区分特征微生物。
常用于权重分析的主要方法包括线性判别分析(LEfSe)、主成分分析(PCA)等(代码5A)。LEfSe是专门针对微生物组数据开发的,其核心功能是用LDA(Fisher,1936年)和MASS(Ripley等,2013年)等包来实现。通过提取PCA排序的loading矩阵,可以找到对样本变化影响最大的微生物作为生物标志物(代码5B)。
在机器学习方面,目前微生物组分析中广泛使用的随机森林模型是通过使用randomforest包(Liaw和Wiener,2002年)来实现(代码5C)。还有许多基于决策树的机器学习模型,例如mboost(Hofner等,2014年)包提供了基于提升算法的方法,e1071(Dimitriadou等,2008年)包提供了支持向量机svm()(代码5D),以及朴素贝叶斯(naiveBayes())。xgboost包可以将多个树模型集成在一起形成强大的分类器,还可以通过多种策略,包括正则化项、Shrinkage and Column Subsampling等,防止过拟合等问题。此外,使用pROC(Robin等,2011年)包用来绘制ROC曲线(代码5D)以评估机器学习模型的效果。Caret包提供交叉验证用来确定特征微生物数量(Kuhn,2008年)。目前,Wirbel等人(2021年)开发了一个开源的R包SIAMCAT,这是一个专门针对微生物组数据的特点进行定制,功能强大且友好的计算机器学习工具包。
相关性和网络分析
微生物共现网络分析是用于寻找可能存在互作关系的微生物模块。共现网络分析主要包括相关性计算、网络可视化和网络属性计算。用于计算相关性的常见R包括psych(Revelle和Revelle,2015年)(代码6A)、WGCNA(Langfelder和Horvath,2008年)(代码6B)、Hmisc(Harrell Jr和Harrell Jr,2019年)(代码6C)和SpiecEasi(Kurtz等,2015年)(代码6D)。在这些R包中,WGCNA具有最高的计算速度,但需要额外的p值矫正;psych可以计算相关和矫正p值,但速度很慢;SpiecEasi包可以使用sparcc方法进行更适合微生物组数据的相关性矩阵计算,并可调用多线程来加速计算。用于网络可视化和属性计算的R包包括igraph(代码6E)、network和ggraph包(代码6F)。这些R包包含许多网络可视化的布局算法。此外,将network包与ggplot2结合实现网络可视化,可以更容易地修改。Sna和ggraph包具有许多可视化布局算法,增加了网络可视化的样式。随着网络分析在微生物组分析中的应用越来越广泛,网络模块化和通过模块寻找关键群体的关注也越来越多。WGCNA包提供了一个完整的框架,可快速完成相关性计算、网络模块计算、模块特征向量计算和其他网络属性探索。最近开发的ggClusterNet(Wen等,2022年)包(代码6G)为微生物组网络提供了一个统一的框架,并设计了多种独特的基于模块的可视化算法,来可视化网络中的模块关系。
功能预测
针对16S rDNA的功能预测,已开发了Tax4Fun(Aßhauer等,2015年)包(代码7A),使用扩增子数据更准确地预测微生物群落功能变化。该包已更新为Tax4Fun2(Wemheuer等,2020年)。Microeco可以实现对细菌/古菌的FAPROTAX(Louca等,2016年)预测和对真菌的FUNGuild(Nguyen等,2016年)预测,该预测是基于精选发表论文中分类功能描述数据库。功能预测可以预测微生物群落功能并进行后续的统计分析。此外,vegan包可用于多样性分析,而edgeR、DESeq2和limma包可用于差异分析。对于功能富集,clusterProfiler(代码7B)包可进行GO、KEGG、GSEA和GSVA富集分析,它考虑了基因/通路的丰度,被推荐使用。此外,clusterProfiler包基于ggplot语法提供绘图功能,可以简单地绘制图形。可以使用WGCNA进行基因/通路网络分析,ggClusterNet进行网络参数计算和可视化。然而,功能预测结果的可靠性,特别是对于环境样品而言,目前存在争议,因此通常需要进一步验证分析结果。
其他微生物组分析
微生物群落形成过程的分析常用Stegen等人(2013)提出的框架,通常结合使用R包minpack.lm、picante、Hmisc、eulerr、FSA、ape、stats4等计算βNTI和RC-Bray指数(代码8A),并推断群落形成过程。Ning等人(2020年)通过基于系统发育分箱的零模型分析定量推断群落构建机制,并开发了R包iCAMP(代码8B)。它可以定量评估不同生态过程(如同质化选择、异质化选择、扩散和漂移)对整个群落和每个系统发育分箱(通常由具有不同生态特征的单科或单目分类群组成)的相对重要性。此外,该包还提供中性理论模型,在群落和分类水平上进行系统发育和分类学零模型分析,计算类群间的生态位差异和系统发育距离,以及检验单个系统发育分箱内的系统发育信号。
微生物群落经常与环境指标进行相关性分分析,例如,使用vegan包提供的mantel.test()来检验微生物群落与环境指标的相关性,使用wascores()、mantel.correlog()来检测微生物群落与环境因素之间的系统发育信号(代码8C)。此外,ggClusterNet包可用于计算微生物/微生物群落与环境因子之间的共现关系,并生成可供发表的图片(代码8D)。
Knights等人(2011)提出了基于R语言的微生物组朔源工具source tracker。Metcalf等(2016)预测了死亡时间并追踪了真实尸体上微生物群落的来源微生物,随后微生物溯源分析逐渐普及。Shenhav等(2019)在R语言中提出了一种新的算法FEAST(代码8E),使微生物溯源分析更加高效快捷,且误报率低。
微生物组集成R包
谢鹏昊翻译,南京农业大学,博士在读;
随着微生物组测序的日益普及,专用于微生物组数据处理的R包也逐渐出现(图2)。McMurdie和Holmes(2013)开发了phyloseq包:这是一个全面的微生物组数据处理工具(包括特征表、系统发育树和特征注解)用于聚类,整合数据导入、存储、分析和输出。该包利用R中的许多工具进行生态和系统发育分析(如vegan, ade4, ape, 和picante),并使用ggplot2来输出高标准的图形。数据存储结构使用类似S4的存储系统将所有相关数据存储为单一的对象,从而使数据共享和分析复现更加容易。随后,microbiome(https://github.com/microbiome/microbiome)
、MicrobiomeAnalystR(Chong等,2020)、microViz(Barnett等,2021)和micreobiomeSeq等包在此框架下出现。接下来,根据R6框架开发的microeco包提供了更多的分析功能。随着数据交互分析的需求,Animalcules(Zhao等,2021)出现了。EasyMicroPlot(https://github.com/xielab2017/EasyMicroPlot)
也使用交互式界面进行微生物组数据的探索,允许快速进行微生物组的下游分析(图3;表1)。
使用Phyloseq包进行微生物组数据分析
Phyloseq利用S4类对象,更适合面向对象的编程,对微生物组数据分析产生了巨大影响(图2/3,图S2A-G, Pipeline 1. phyloseq.Rmd)。通过S4类对象,phyloseq允许数据的五个部分(特征表、注释信息、分组信息、代表性序列和进化树)在同一框架下保持对应,并提供了关于微生物特征和样本的多种过滤功能,允许五部分数据在不考虑数据差异的情况下保持一致的过滤。它还通过微生物数据过滤和标准化,提供了微生物组多种分析,包括多样性计算(图S2A-B),微生物组成可视化(图S2C-D),进化树可视化,和网络分析(图S2E)。其Beta多样性功能提供了超过30种距离算法,远超过vegan等包提供的。其次,phyloseq包使用ggplot进行图形可视化(图S2F),这使得生成和修改图形变得更加容易。此外,phyloseq可以在树的枝和叶上整合进化树和物种分类和丰度(图S2G),使得树信息丰富且美观。
使用microbiome包进行微生物组数据分析
microbiome包也像phyloseq一样使用S4类对象,也可以执行大部分微生物组的分析(图2/3,图S3A-G, Pipeline 2. Microbiome.Rmd)。它包括微生物多样性分析(图S3A-E),以及差异分析(图S3F-G)。与phyloseq相比,microbiome包在alpha多样性指标上更为丰富,提供了超过30种alpha多样性指标。其次,它提供了核心微生物计算和可视化功能。总的来说,它可以作为对phyloseq的补充,或者与之联合使用。
使用MicrobiomeAnalystR包进行微生物组数据分析
MicrobiomeAnalystR是MicrobiomeAnalyst在线版本的R包(图2/3,图S4A-J, Pipeline 3. MicrobiomeAnalystR.Rmd)。包括多样性分析(图S4A-F)、差异分析(图S4G)、生物标记识别(图S4H-I)以及样本测序库大小概览(图S4J),这些功能比前两个包更强大。在可视化方面,它结合了基础包、ggplot绘图以及交互式绘图。在网络分析方面,它提供了计算和绘制更适合微生物组数据的SparCC网络的过程。然而,这个包依赖于许多来自CRAN、Bioconductor和GitHub的R包,因此完全安装MicrobiomeAnalystR需要付出大量的努力。
使用Animalcules包进行微生物组数据分析
Animalcules包是在交互式平台上进行分析的一种方式(图2/3,图S5A-J, Pipeline 4. Animalcules.Rmd)。它可以对样本进行基础信息统计与绘图(图S5A)或交互式饼图(图S5B),计算和可视化alpha多样性(图S5C),微生物组成或功能组成的热图和堆积柱状图(图S5D-E),特征丰度箱线图(图S5F),属水平的bray距离热图(图S5G),排序分析(图S5H-I),使用随机森林、逻辑回归选择生物标记(图S5J)等其他分析。这些分析的结果通常可以通过交互式修改参数进行再分析,图片可以进行交互式的放大和缩小,点击查看详细信息,以及由鼠标进行的其他操作以更好地修改。然而,结果不能导出为矢量格式,这并不满足出版的要求。其次,分析内容太少,特别是微生物组网络分析,微生物组与其他指标之间的相关性分析。
使用microeco包进行微生物组数据分析
microeco包非常强大,使用R6类数据结构(图2/3,图S6A-L, Pipeline 5. microeco.Rmd)。它包括微生物多样性(图S6A/B)物种组成(图S6C-E),差异分析(图S6F-H),生物标记物寻找(图S6I-J),网络分析(图S6K),以环境因子探索(图S6L),以及系统发育多样性分析等。它几乎可以完成目前所有的微生物组分析内容。然而,它并不适合初学者使用,因为使用S6类对象有一定的门槛。另外,由于功能过多,对输入数据的要求不同,导致一些功能难以使用。
使用amplicon包进行微生物组数据分析
amplicon包是微生物组分析工具包EasyMicrobiome(Liu等,2023)中的一个分析和绘图工具(图2/3,图S7A-I,Pipeline 6. Amplicon.Rmd)。它能够进行各种多样性分析,包括alpha多样性(图S7A),稀化曲线(图S7B),聚类距离热图(图S7C)和PCoA(图S7D),NMDS,LDA和PCA,物种组成(图S7E/F),差异分析(图S7G/H)。它可以轻松生成高质量的图像,如用于多样性分析的箱线图,散点图,用于分类或功能组成的堆叠条形图,环形图和maptree图。它的一个显著特点是能够精细调整图像的呈现,从而生成可以发布的图像。此外,amplicon包内的几个工具可用于微生物组数据的转换,有助于使用LEfSe和STAMP等工具进行后续分析。然而,在当前版本,amplicon包并未提供网络分析,微生物组-环境相互作用分析,以及群落形成过程分析的一些功能。作者在EasyAmplicon流程中提供了一些脚本来实现这些功能,并在发表的论文中提到计划在未来完成这些功能。。
图2. 微生物分析集成R包的功能介绍
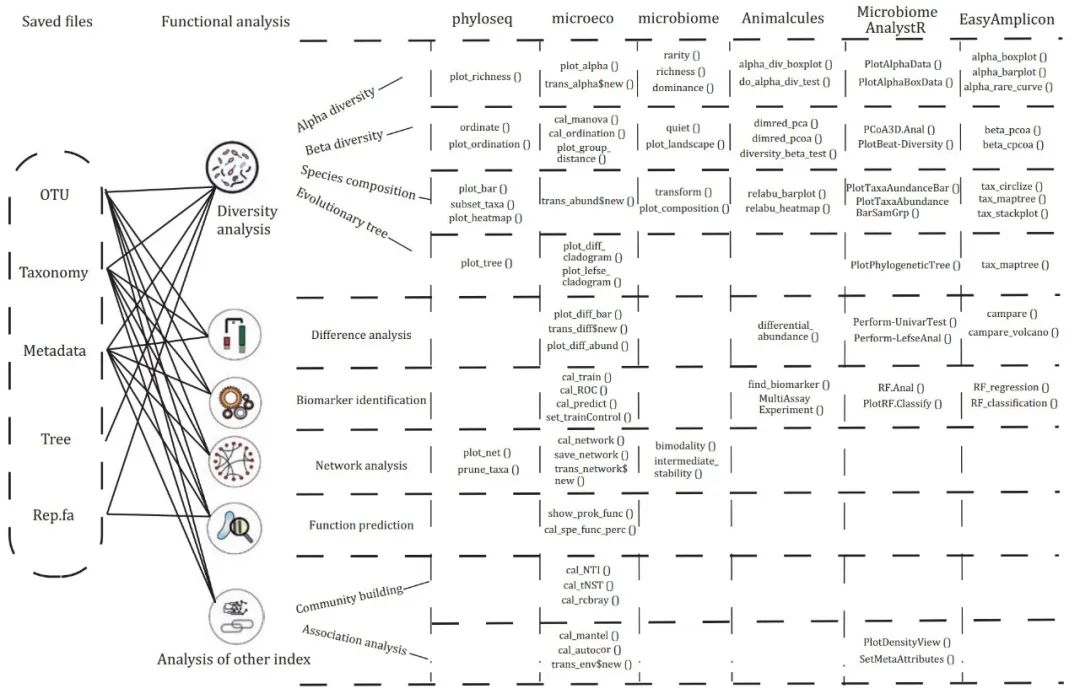
微生物群落分析可分为多样性 分析、差异分析、生物标志物鉴定、相关和网络分析、功能预测、以及其他微生物群落分析(群落建立/构建过程,与其他指标的关联分析)。
图3. 微生物群落分析集成R包的典型结果和类似结果的比较
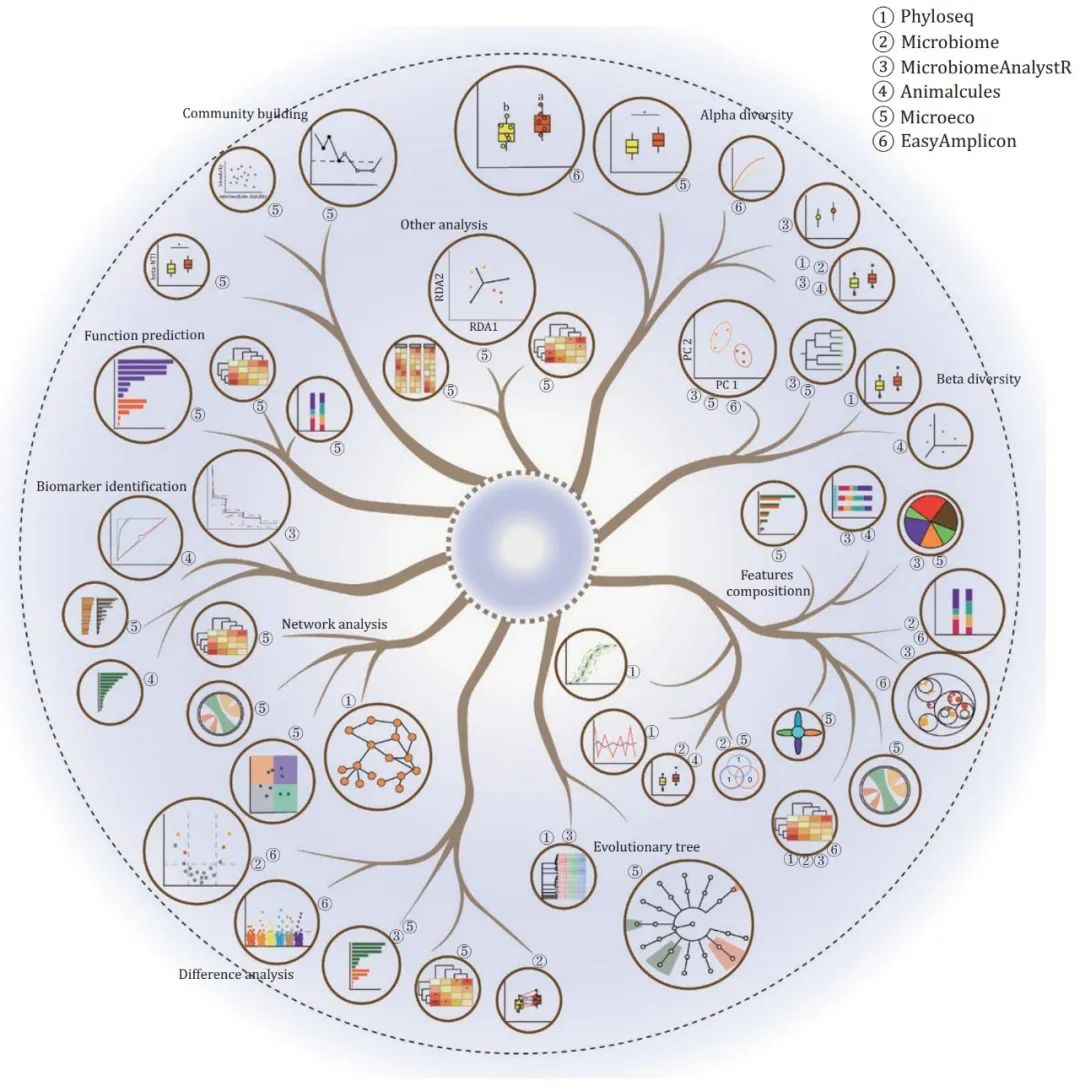
根据微生物群落分析功能的主要类别,对多个集成R包的分析结果进行分组。树状图中的每个主要分支 代表微生物群落分析的一种类型,共有10个主要分支:特征多样性分析,包括(i)alpha多样性 分析;(ii)beta多样性分析;(iii)特征(群落分类或功能)组成分析;(iv)进化或分类树分析;(v)差异分析;(vi)生物标志物鉴定;(vii)相关和网络分析;(viii)功能预测;(ix)群落形成/构建过程分析;(x)其他分析,如与其他指标的关联分析。每片叶子(圆圈)代表分析结果的一种风格样式,叶子外面的圆圈号码代表分析结果所来自的集成R包编号。
基于R语言的微生物组数据挖掘最佳流程
谢鹏昊,牛国庆翻译,南京农业大学,博士在读;
过多的R包可能会阻碍微生物组研究人员有效地选择适合微生物组相关分析的R包。因此,我们在六种分析中选择了高效、常用、用户友好的函数(图S8):1) 多样性分析(图S9A-I; 图S10A-E),2) 差异分析(图S10F-I; 图S11A-B),3) 生物标志物识别(图S11C-D),4) 相关性和网络分析(图S11E-I),5) 功能预测,6) 其他微生物组分析(图S12A-I)。所有脚本都可以在Pipeline.BestPractice.Rmd文件中找到。
在这个流程中,我们使用amplicon包进行Alpha多样性稀疏曲线(图4A;图S9A)和PCoA分析(图4B;图S9B),使用ggplot2包可视化微生物群落组成,使用ggClusterNet构建Venn网络(Chen等,2021年)(图4C),使用ggtree和ggtrextre构建进化树(图4D),使用LEfSe生成 cladograms(图4E)。我们使用stst4、ggplot2和cowplot包进行差异分析并生成STAMP图(图4F),使用edgeR进行差异分析并在曼哈顿图中进行可视化(图4G),并应用DESeq2进行差异分析并生成多组火山图(图4H)。我们还使用el071、caret、randomforest和ROC包进行各种机器学习分析并生成特征微生物重要性加权图(图4I)。此外,我们还使用ggClusterNet进行微生物网络分析(图4J),构建网络图和绘制组合图以探究环境因素和微生物群落之间的关系(图4K)。最后,我们使用FEAST包进行群落溯源分析并构建饼图(图4L)。其他分析包括微生物群落组成的堆积柱状图(图S9E/H)、和弦图(图S10A)、Venn图(图S10C)、Upset图(图S10D)、差异分析火山图(图S10F)、功能预测等。
图4. R语言中微生物群落分析的最佳实践结果实例
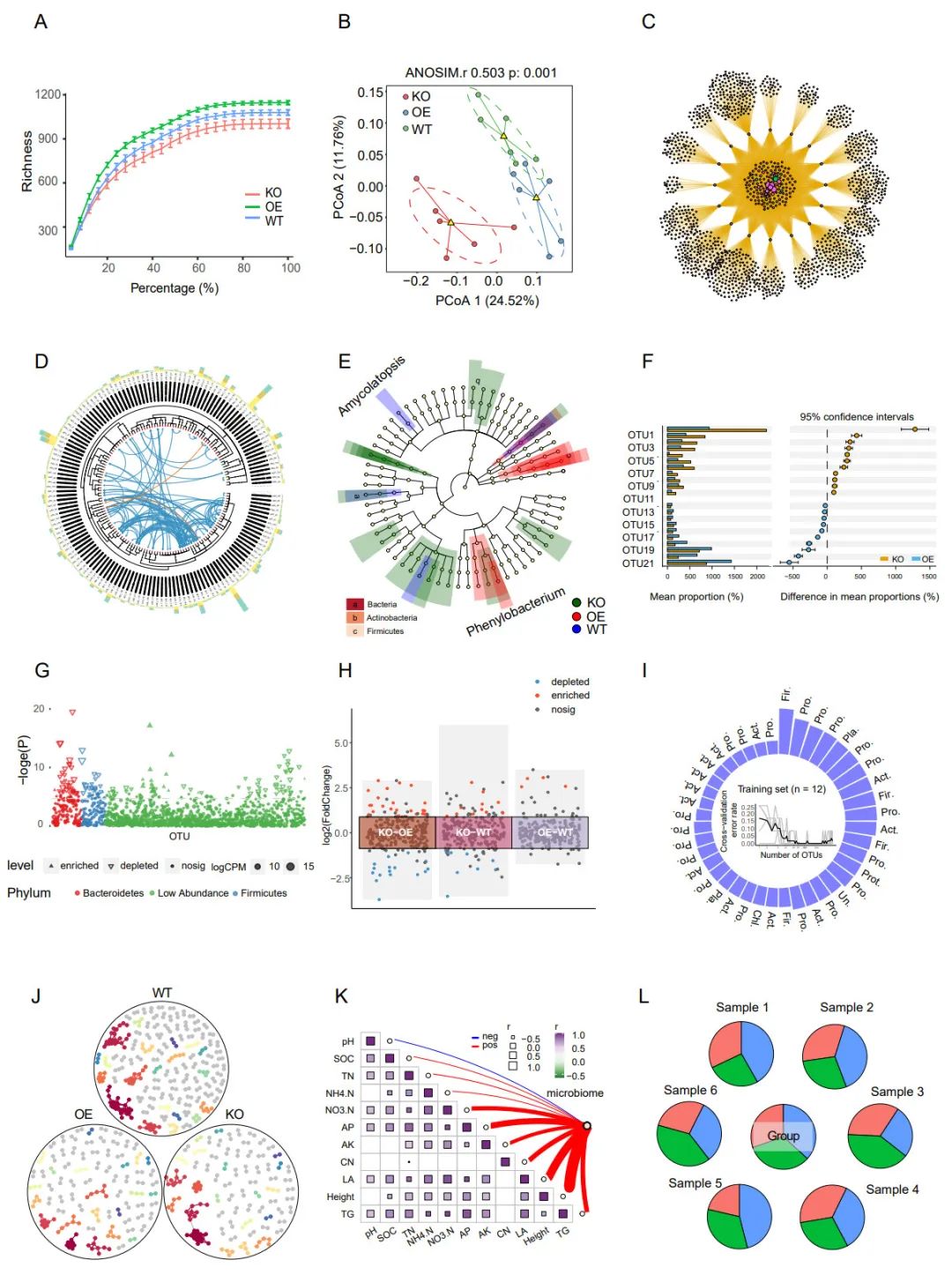
所选结果包括稀疏曲线(A)、
主坐标分析散点图(B),维恩网络图(C),进化树(D),LEfSe图(E),STAMP的差异分析误差条图(F),差异分析曼哈顿图(G),差异分析多组火山图(H),生物标志物选择环列图 (I),网络图(J),组合图(K),溯源分析饼图(L)
总结与展望
在过去的十年中,R语言和众多R包在微生物组数据分析中发挥了重要作用。R语言易于使用和入门,吸引了许多研究人员学习使用。然而,在微生物组数据分析中,供给和需求之间仍存在一些矛盾。例如,在Windows系统下支持多线程往往很困难;其次,许多R包的运行速度相对较慢;第三,R包在微生物组应用中仍需要进一步发展。例如,缺乏允许探索基于时间序列的微生物组成的软件包,以及更强大的交互式软件包来分析复杂的微生物数据。此外,ggplot2缺乏创建复杂和组合图形的能力,无法满足多个复杂指标与微生物群落数据之间关系的可视化要求。因此,有必要开发新的R包以更适合绘制复杂图形和组合图形。
随着测序技术的发展,数据分析方法也随着R包的发展而在微生物组领域取得了进步。这些R软件包从经典的R包如vegan,到集成的R包如phyloseq(包含了许多功能,并设置了统一的数据处理框架,已经能够实现微生物组分析的大多数功能,包括微生物多样性、差异、生物标志物鉴定、相关性和网络、系统发育分析等)。然而,这些R包存在一些冗余的功能;例如,phyloseq、microbiome等可以进行微生物多样性分析,差异仅在于可视化方法和方案。这种类似的情况在微生物组分析的R软件包中一直存在,因此我们希望在未来的开发中,尽量避免重复使用相同部分或类似内容,以突出R包的优势。
虽然这些R包可以实现很多功能,但在一些具体的分析中可能存在不足,比如在Alpha和Beta多样性分析中,图形往往没有展示差异检验结果,难以直观地从图形中观察出差异。此外,还有一些内容需要进一步开发,例如将更多的机器学习方法应用于微生物组数据,包括学习方法、模型和重要变量的评估等。其次,宏基因组的应用越来越广泛,基于Kraken(Wood和Salzberg,2014)、MEGAN(Huson等,2007)、MetaPhlAn2(Truong等,2015)、HUMAnN2(Franzosa等,2018)、eggNOG-mapper(Huerta-Cepas等,2017)等物种和功能注释的结果从兆字节(M)升级到千兆字节(G)。因此,应使用更快速的数据处理R包来进行微生物组数据分析,例如data.table、fst、tidyfst等。
使用适当的数据结构可以加速微生物组数据分析。起初,我们使用S4类对象进行微生物组数据封装,可以全面有效地完成各种分析。R6类对象和其他对象的出现极大地影响了微生物组数据的处理,并在很大程度上促进了数据的处理。随着R语言tidy家族的发展,最近出现了基于tidy的数据结构,用于微生物组数据挖掘。例如,MicrobiotaProcess包(Xu等,2023)。这种结构更适用于微生物组数据挖掘、机器学习建模等分析,在分析中可以更容易提取实验设计、时间、空间等因素对微生物组数据的影响,发现深层次的规律。我们期待R语言能使微生物组分析更加高效,帮助大家发现更多关于微生物在人类、动物、植物和环境中的作用,并利用微生物为我们造福,使世界变得更加美好。
翻译:杨盛蝶/牛国庆/谢鹏昊,南京农业大学,博士在读
校对:文涛,南京农业大学,博士后
审核:刘永鑫,中国农业科学院深圳农业基因组研究所,研究员
引文格式
Tao Wen, Guoqing Niu, Tong Chen, Qirong Shen, Jun Yuan, Yong-Xin Liu. 2023. The best practice for microbiome analysis using R. Protein & Cell pwad024. https://doi.org/10.1093/procel/pwad024
猜你喜欢
iMeta简介 高引文章 高颜值绘图imageGP 网络分析iNAP
iMeta网页工具 代谢组MetOrigin 美吉云乳酸化预测DeepKla
iMeta综述 肠菌菌群 植物菌群 口腔菌群 蛋白质结构预测
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature
系列教程:微生物组入门 Biostar 微生物组 宏基因组
专业技能:学术图表 高分文章 生信宝典 不可或缺的人
一文读懂:宏基因组 寄生虫益处 进化树 必备技能:提问 搜索 Endnote
扩增子分析:图表解读 分析流程 统计绘图
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流快速解决科研困难,我们建立了“宏基因组”讨论群,己有国内外6000+ 科研人员加入。请添加主编微信meta-genomics带你入群,务必备注“姓名-单位-研究方向-职称/年级”。高级职称请注明身份,另有海内外微生物PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。
点击阅读原文



