使用rWCVP在WCVP中匹配名称
- 加载库
- 工作流
- 1. 示例数据:IUCN红色名录
- 2. 将匹配的名称解析为接受名
- 2.1 模糊匹配
- 2.2 多项匹配
- 2.3 将评估与接受的名称相关联
- 3. 可视化匹配过程
- 4. 得到最终数据集
加载库
世界维管植物名录提供了所有已知维管植物物种的全球共识观点,包括分类地位和同义词。因此,它可用于标准化和协调来自其他来源的植物名称列表。
rWCVP 中实现了名称匹配功能,使用户更容易根据 WCVP 标准化植物名称列表。
在本文中,我们将向您展示一个例子,说明rWCVP中的名称匹配如何适应实际的工作流。
除了rWCVP,我们还将使用tidyverse软件包集合进行数据操作,gt用于格式化表格,ggallaul用于可视化匹配过程。
library(rWCVP)
library(tidyverse)
library(gt)
library(ggalluvial)
工作流
1. 示例数据:IUCN红色名录
我们在这里使用的例子是将IUCN濒危物种红色名录(红色名录)中的评估与WCVP中认可的名称进行匹配。
我们将使用2022-1版红色名单中的植物评估下载
redlist = read_csv("D:/ALL_Softwares/R-4.2.0/library/rWCVP/extdata/redlist-summary_2022-1.csv",col_types = cols(.default = col_character()))
glimpse(redlist)
Rows: 61,015
Columns: 3
$ scientific_name <chr> "Cotoneaster granatensis", "Juniperus drup…
$ authority <chr> "Boiss.", "Labill.", "(Pancic) Purk.", "Bo…
$ category <chr> "LR/cd", "LC", "EN", "VU", "LC", "EN", "VU…
现在我们已经从 Red List 加载了名称,我们可以使用 wcvp_match_names 函数将它们与 WCVP 匹配。
此函数采用名称数据框进行匹配,数据框中存储名称的列的名称,以及(可选)存储每个名称作者的列的名称,如果要将其包含在匹配过程中.
该函数将首先尝试查找与 WCVP 中的名称完全匹配的任何名称。如果提供了作者列,则第一步将包括作者,第二步将运行以完全匹配没有作者字符串的任何剩余名称。所有剩余的不匹配的名称然后通过模糊匹配过程,该过程尝试在语音上匹配名称,然后通过 Levenshtein 距离找到最相似的名称。
matches = wcvp_match_names(redlist,name_col = "scientific_name",author_col = "authority",fuzzy = TRUE,progress_bar = FALSE)
── Matching names to WCVP ──────────────────────────────────────────
ℹ Using the `scientific_name` column ── Exact matching 61015 names ── ✔ Found 60165 of 61015 names ── Fuzzy matching 850 names ── ✔ Found 826 of 850 names ── Matching complete! ── ✔ Matched 60928 of 61015 names
ℹ Exact (with author): 43268
ℹ Exact (without author): 16897
ℹ Fuzzy (edit distance): 398
ℹ Fuzzy (phonetic): 365
! Names with multiple matches: 391
Matching ■■■■■■■■■■■■■■■■■■■■■■■■■■■■■■■ 100% | ETA: 4s
我们还没有显式地传入 WCVP 名称的数据框,并且我们收到警告,因为我们正在通过 rWCVPdata 包使用过时的版本。我们现在可以忽略此警告,因为该包(以及本教程)是使用网站上当前未提供的数据版本开发的。
我们还收到了一系列信息,让我们知道精确匹配的比例,以及模糊匹配需要多长时间。
完成后,我们将获得一个完整的摘要,其中包含匹配的名称数量、使用模糊匹配的数量以及与 WCVP 中的多个名称匹配的数量。
wcvp_match_names 的输出是我们原始名称的数据集,它们与 WCVP 中的哪些名称匹配,以及有关它们如何匹配以及任何模糊匹配的接近程度的信息。
2. 将匹配的名称解析为接受名
现在我们已经匹配了我们的名称,我们可以解决模糊匹配和与 WCVP 中的多个条目匹配的名称,并确保我们评估的物种与 WCVP 中接受的名称相关联。
如何选择哪些模糊匹配是有效的,以及如何解决多个匹配,最终将取决于您进行匹配的原因。例如,我们将 IUCN 评估与 WCVP 中接受的名称进行匹配。对于此应用中,评估仅对特定的分类概念有效,因此我们可能不关心解析与非同型同义词匹配的任何内容。
在本教程中,在过滤掉不适合我们的应用的匹配之前,我们首先尝试解决尽可能多的模糊和多重匹配。
2.1 模糊匹配
模糊匹配不多(大约 1000 个),但仍有很多需要手动验证,所以让我们根据以下规则进行一些预检查:
- 手动检查所有相似度低于90%的匹配
- 如果模糊匹配具有相同的作者字符串并且相似度≥90%,我们将保留它。
- 如果模糊匹配只差一个字母(即编辑距离为1)并且相似度≥90%,我们保留它。
fuzzy_matchs = matches %>%filter(str_detect(match_type, "Fuzzy")) %>%mutate(keep = case_when(match_similarity < 0.9 ~ NA_real_,wcvp_author_edit_distance == 0~1,match_edit_distance == 1 ~ 1))
table(fuzzy_matchs$keep, useNA = "always")
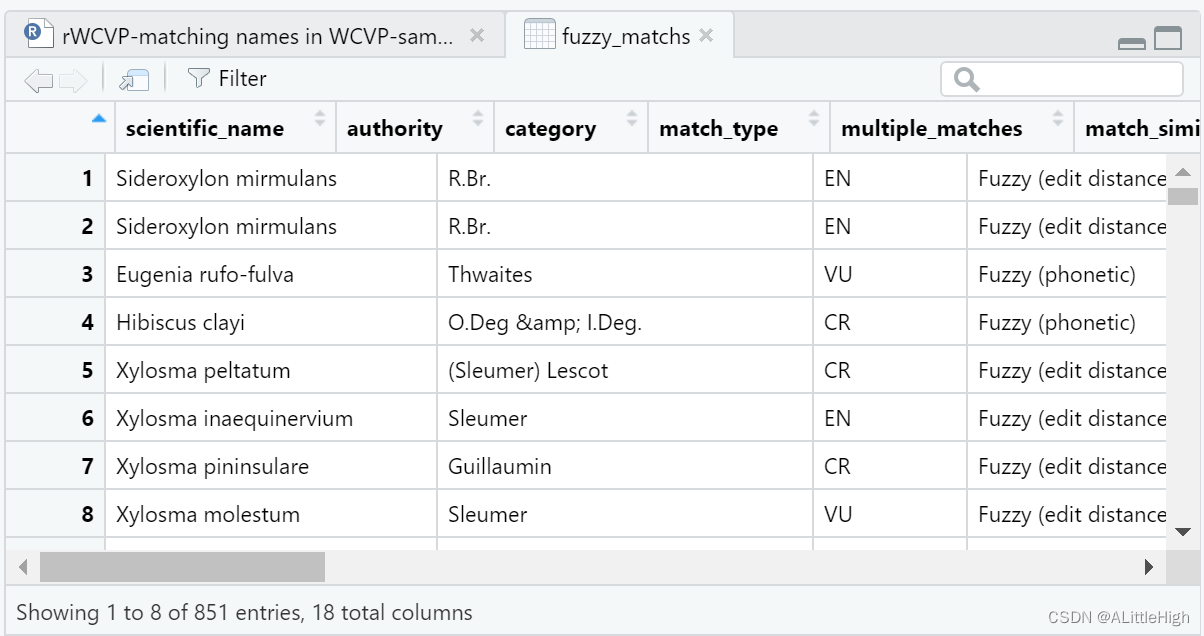
1 <NA> 534 317
超过一半的模糊匹配无需查看即可解决。
编辑距离为 2 怎么样——这肯定还是很接近吧?
fuzzy_matchs %>%filter(match_edit_distance == 2,!multiple_matches,match_similarity > 0.9) %>%arrange(desc(wcvp_author_edit_distance)) %>% select(scientific_name, authority, match_type, wcvp_name, wcvp_authors) %>%head()
# A tibble: 6 × 5scientific_name authority match_type wcvp_name wcvp_authors<chr> <chr> <chr> <chr> <chr>
1 Diospyros tampolensis H.N.Rako… Fuzzy (ed… Diospyro… H.Perrier
2 Diospyros crassifolia A.G.Lina… Fuzzy (ed… Diospyro… D.Don
3 Diospyros nitidifolia A.G.Lina… Fuzzy (ed… Diospyro… Elmer
4 Garcinia eugeniaefolia Wall Fuzzy (ph… Garcinia… (Choisy) Wa…
5 Hebepetalum humiriifo… (Planch.… Fuzzy (ed… Hebepeta… (Planch.) B…
6 Diospyros ambanjensis G.E.Scha… Fuzzy (ed… Diospyro… Gürke
显然不是!
如果我们真的想从算法上继续,我们可以随机抽取100个名字,测试各种规则的准确性——关于使用相似性的示例,请参见Nic Lughadha等人(2020年)的支持信息。
然而,在这一点上,快速浏览每个名字会给出最准确的结果,而且速度会更快。
write_csv(fuzzy_matchs, "redlist-fuzzy-tocheck.csv")
在Excel中浏览这些花了不到一个小时;我在keep列中用1标记好的匹配,用0标记坏的匹配。然后,我删除了除不良匹配的match_type和multiple_matches之外的所有匹配数据,并添加了一个新列resolved_match_type。我为好的匹配留了空白,为差的匹配填了“模糊匹配被拒绝”。
fuzzy_checked = read_csv("D:/ALL_Softwares/R-4.2.0/library/rWCVP/extdata/redlist-fuzzy-checked.csv",show_col_types = FALSE) %>%select(-keep) %>%mutate(resolved_match_type=ifelse(! is.na(resolved_match_type),resolved_match_type,match_type))
checked_matches = matches %>%filter(! str_detect(match_type, "Fuzzy")) %>%bind_rows(fuzzy_checked)
到目前为止,我们保持了相同的行数——我们还不想去掉不匹配的行。
2.2 多项匹配
现在我们需要处理多个匹配项。同样,我们可以使用一些规则来自动解决这些问题:
- 使用作者信息过滤匹配项。如果一个或多个匹配项具有相同的作者字符串,我们将保留它们。
- 如果一个(且只有一个)匹配项被接受,我们将保留该匹配项。
- 如果一个(并且只有一个)匹配项是同义词(与无效、非法等相反),我们将保留那个匹配项。
我们将把这些规则写入一个函数。
resolve_multi = function(df) {if (nrow(df) == 1) {return(df)}# some fuzzy matches are rejected from the previous sectionvalid_matches = filter(df, !is.na(match_similarity))if (nrow(valid_matches) == 0) {return(head(df, 1))}matching_authors =valid_matches %>%filter(wcvp_author_edit_distance == 0 | ! sum(wcvp_author_edit_distance == 0,na.rm=TRUE))if (nrow(matching_authors) == 1) {return(matching_authors)}accepted_names =matching_authors %>%filter(wcvp_status == "Accepted" | ! sum(wcvp_status == "Accepted"))if (nrow(accepted_names) == 1) {return(accepted_names)}synonym_codes = c("Synonym", "Orthographic", "Artificial Hybrid", "Unplaced")synonyms =accepted_names %>%filter(wcvp_status %in% synonym_codes | ! sum(wcvp_status %in% synonym_codes))if (nrow(synonyms) == 1) {return(synonyms)}n_matches = length(unique(synonyms$wcvp_accepted_id)) / nrow(synonyms)final = head(synonyms, 1)if (n_matches != 1) {final =final %>%mutate(across(wcvp_id:resolved_match_type & where(is.numeric), ~NA_real_),across(wcvp_id:resolved_match_type & where(is.character), ~NA_character_),resolved_match_type="Could not resolve multiple matches")}final
}
现在我们遍历具有多个匹配项的每个名称并应用这些规则。
auto_resolved =checked_matches %>%nest_by(scientific_name) %>%mutate(data=list(resolve_multi(data))) %>%unnest(col=data) %>%ungroup()auto_resolved =auto_resolved %>%mutate(resolved_match_type=case_when(is.na(resolved_match_type) & is.na(match_type) ~ "No match found",is.na(resolved_match_type) ~ match_type,TRUE ~ resolved_match_type))count(auto_resolved, resolved_match_type)
# A tibble: 6 × 2resolved_match_type n<chr> <int>
1 Could not resolve multiple matches 6
2 Exact (with author) 43266
3 Exact (without author) 16893
4 Fuzzy (edit distance) 323
5 Fuzzy (phonetic) 350
6 Fuzzy match rejected 90
我们仍然找不到匹配的约 90 个名称(不到原始数据集的 1%)。对于大多数数据集,这是一个可以接受的损失,但这个特定数据将用于多篇论文,因此值得更仔细地研究。
auto_resolved %>%filter(resolved_match_type %in% c("No match found","Fuzzy match rejected")) %>%write_csv("redlist_tomanuallymatch.csv")
manually_resolved = read_csv("D:/ALL_Softwares/R-4.2.0/library/rWCVP/extdata/redlist-manually-matched.csv",show_col_types=FALSE)
count(manually_resolved, resolved_match_type)
# A tibble: 3 × 2resolved_match_type n<chr> <int>
1 Manually matched 14
2 New/undescribed species 55
3 No valid match found 21
在这个例子中,许多不匹配的名称是没有被添加到WCVP的新物种(有时被列为例如Heptapleurum sp。),比单独搜索每个名字更快。尽管如此,这仍然是一个耗时的过程,成功率相对较低(这里,我们找到了95个名字),因此它不适用于所有(甚至大多数)名称匹配工作流。
我们需要对手动匹配的名称重新运行匹配,以获取其余信息。
manually_resolved =manually_resolved %>%wcvp_match_names(name_col = "manually_entered_name", fuzzy=FALSE)
── Matching names to WCVP ───────────────────────────────────────────────
ℹ Using the `manually_entered_name` column
! No author information supplied - matching on taxon name only── Exact matching names ── ✔ Found 13 of names ── Matching complete! ── ✔ Matched 13 of 15 names
ℹ Exact (without author): 13
ℹ No match found: 2
! Names with multiple matches: 0
Matching ■■■■■■■■■■■■■■■■■■■■■■■■■■■■■■■ 100% | ETA: 3s
这又产生了几个多重匹配。我们可以通过输入我们想要的WCVP ID而不是名称来避免这种情况。但在这种情况下,它只影响几个记录。看看这些,额外的名字是不合法的,所以我们可以快速过滤掉。
manually_resolved = filter(manually_resolved, wcvp_status != "Illegitimate")
现在,我们将它们重新插入到匹配结果中,并删除任何不匹配的内容。
resolved_matches =manually_resolved %>%select(-c(manually_entered_name, Notes, match_type, multiple_matches,match_similarity, match_edit_distance)) %>%rename(match_type=original_match_type) %>%bind_rows(auto_resolved %>%filter(! scientific_name %in% manually_resolved$scientific_name))
最后,我们将在一个格式良好的表格中查看名称解析的摘要。
resolved_matches %>%count(resolved_match_type, sort=TRUE) %>%gt(rowname_col = "resolved_match_type") %>%tab_options(table_body.hlines.color = "transparent",column_labels.hidden = TRUE) %>%summary_rows(columns = c(n),fns = list(Total ="sum"),formatter = fmt_integer) %>%tab_style(style = list(cell_text(align = "left")),locations = cells_stub()) %>%tab_header("Match summary", "(after resolution of fuzzy and multiple matches)") %>%opt_align_table_header(align = "left")
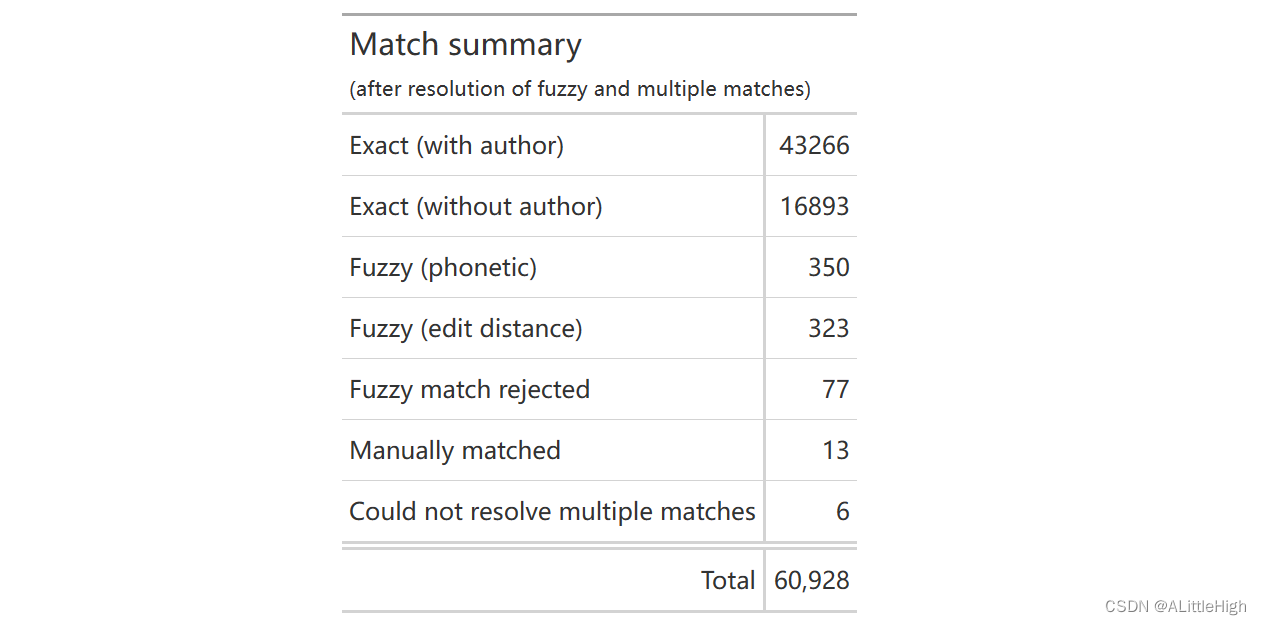
2.3 将评估与接受的名称相关联
在我们完成名称匹配之前还有最后一步——将每个红色名录评估链接到一个可接受的物种名称。这是事情变得有点棘手的地方 - 对于同义词,我们需要考虑名称更改如何影响评估。具体来说,只有在保留分类概念的情况下,我们才能将新名称用于评估。
幸运的是,我们可以使用 WCVP 的 homotypic_synonym 列来轻松过滤我们的匹配项。
我们会将完整的 WCVP 添加到我们的数据中,使用 wcvp_accepted_id 与 plant_name_id 连接。然后,我们只想标记与已接受物种匹配或与链接到已接受物种的同型同义词匹配的名称。
accepted_matches = resolved_matches %>%left_join(rWCVPdata::wcvp_names, by=c("wcvp_accepted_id"="plant_name_id")) %>%mutate(keep=case_when(taxon_status == "Accepted" & (wcvp_status != "Synonym" | wcvp_homotypic) ~"Matched to an accepted name",TRUE ~ "Not matched to an accepted name"))count(accepted_matches, keep)
# A tibble: 2 × 2keep n<chr> <int>
1 Matched to an accepted name 59084
2 Not matched to an accepted name 1844
3. 可视化匹配过程
我们可能想做的一件事是查看匹配过程的进展情况,即可视化经历每种类型匹配的姓名比例。冲积图提供了一种方法。
step1_codes = c("Exact (with author)"="Exact","Exact (without author)"="Exact","Fuzzy (edit distance)"="Fuzzy","Fuzzy (phonetic)"="Fuzzy")
step2_codes = c("Exact (without author)"="\U2713","Exact (with author)"="\U2713","Fuzzy (edit distance)"="\U2713","Fuzzy (phonetic)"="\U2713","Could not resolve multiple matches"="\U2716","Fuzzy match rejected"="\U2716","No match found"="\U2716")
plot_data =accepted_matches %>%mutate(step0="Input",step1=recode(match_type, !!! step1_codes),step2=recode(resolved_match_type, !!! step2_codes),step3=ifelse(keep == "Matched to an accepted name", "\U2713", "\U2716")) %>%replace_na(list(multiple_matches=FALSE)) %>%mutate(scenario=paste0(step1, step2, step3)) %>%count(step0, step1, step2, step3, scenario) %>%mutate(colour_key=step3) %>%to_lodes_form(axes=c(1:4), id="scenario") %>%group_by(x, stratum) %>%mutate(label=ifelse(row_number() == 1, as.character(stratum), NA_character_),total=sum(n)) %>%ungroup() %>%mutate(label=ifelse(total < 1500, NA_character_, label))plot_data %>%ggplot(mapping=aes(x=x, y=n, stratum=stratum, alluvium=scenario, fill=colour_key)) +scale_fill_brewer(palette="Set2") +geom_flow(stat="alluvium", lode.guidance="frontback", color="darkgrey",aes.flow="forward") +geom_stratum() +geom_text(mapping=aes(label=label), vjust=0.75, size=4) +annotate("text", x=c(2, 3, 4), y=rep(61371 * 1.03, 3),label=c("Initial", "Resolved", "Accepted"), size=5) +guides(fill="none") +theme_void()
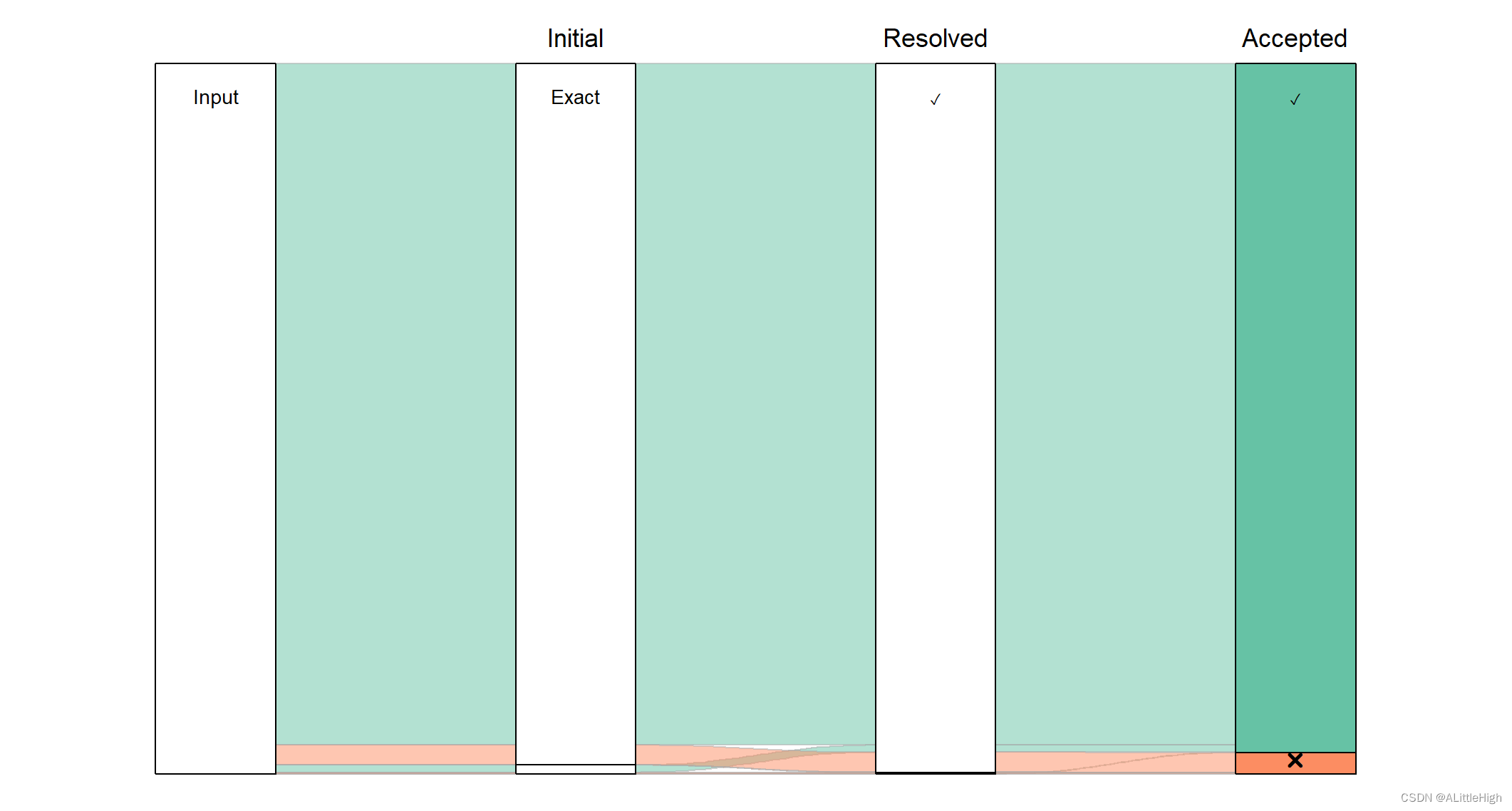
显然,绝大多数名称都有完全匹配(有或没有作者字符串),不需要解析。如果我们将它们排除在外,有效地放大该图的下部,我们可以看到不太直接的匹配的更多细节。
plot_data =accepted_matches %>%mutate(step1=str_replace(match_type, "\\(", "\n\\("),step2=str_replace(resolved_match_type, "\\(", "\n\\("),step3=ifelse(keep == "Matched to an accepted name", "\U2713", "\U2716")) %>%mutate(scenario=paste0(step1, step2, step3)) %>%count(step1, step2, step3, scenario) %>%mutate(colour_key=step3) %>%to_lodes_form(axes=c(1:3), id="scenario") %>%mutate(label1=ifelse(stratum %in% c("\U2713", "\U2716"),as.character(stratum), NA_character_),label2=ifelse(! stratum %in% c("\U2713", "\U2716"),as.character(stratum), NA_character_))plot_data %>%#filter out the big categoriesfilter(n < 4000) %>%ggplot(aes(x=x, y=n, stratum=stratum, alluvium=scenario, fill=colour_key)) +scale_fill_brewer(palette = "Set2") +geom_flow(stat="alluvium", lode.guidance="frontback",color="darkgray", aes.flow="forward") +geom_stratum() +theme_void()+geom_text(mapping=aes(label=label1), stat="stratum", size=8) +geom_text(mapping=aes(label=label2), stat = "stratum", size=4) +annotate("text", x=c(1, 2, 3), y=2950, label=c("Initial", "Resolved", "Accepted")) +annotate("text", x=3.4, y=1000,label="e.g. heterotypic \nsynonyms, \nunplaced names", size=4) +theme(legend.position = "none")

4. 得到最终数据集
最后,我们希望通过过滤掉所有不成功的匹配并减少列的数量(并对它们进行重命名以使其更加直观),将我们的大型数据框架转换成更易于管理的东西,以便我们可以用于下游分析。
final_matches =accepted_matches %>%filter(keep == "Matched to an accepted name") %>%select(scientific_name, authority, category,match_name=wcvp_name, match_status=wcvp_status,accepted_plant_name_id=wcvp_accepted_id, ipni_id,accepted_taxon_name=taxon_name, accepted_taxon_authors=taxon_authors)glimpse(final_matches)
Rows: 59,084
Columns: 9
$ scientific_name <chr> "Actinodaphne leiantha", "Camellia dongn…
$ authority <chr> "Hook.f.", "Orel", "Nees", "Lapeyr.", "(…
$ category <chr> "DD", "CR", "LC", "LC", "LC", "LC", "LC"…
$ match_name <chr> "Actinodaphne leiophylla", "Camellia don…
$ match_status <chr> "Accepted", "Accepted", "Accepted", "Acc…
$ accepted_plant_name_id <dbl> 2620909, 2694538, 225431, 228432, 278344…
$ ipni_id <chr> "462276-1", "60443091-2", "298997-1", "3…
$ accepted_taxon_name <chr> "Actinodaphne leiophylla", "Camellia don…
$ accepted_taxon_authors <chr> "(Kurz) Hook.f.", "Orel", "L.", "Lapeyr.…
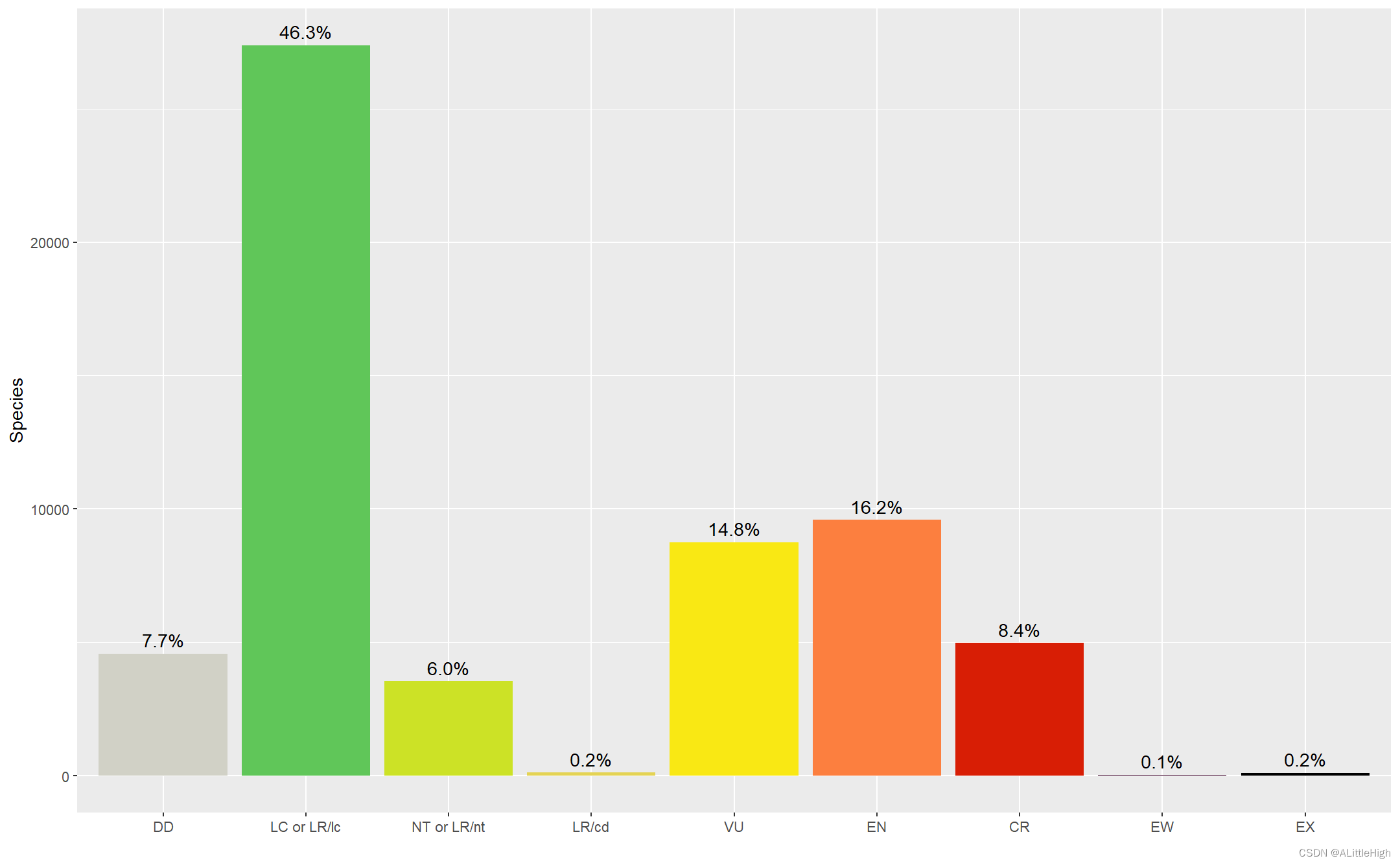
此外,作为下游分析的一个例子,我们可能希望显示每个红色列表类别中被接受的物种的数量。
cat_order = c("DD", "LC or LR/lc", "NT or LR/nt", "LR/cd", "VU", "EN", "CR","EW", "EX")
cat_colors = c("DD"="#D1D1C6", "LC or LR/lc"="#60C659", "NT or LR/nt"="#CCE226","LR/cd"="#e4d354", "VU"="#F9E814", "EN"="#FC7F3F", "CR"="#D81E05","EW"="#542344", "EX"="#000000")final_matches %>%mutate(category=recode(category, "LC"="LC or LR/lc", "LR/lc"="LC or LR/lc","NT"="NT or LR/nt", "LR/nt"="NT or LR/nt"),category=factor(category, levels=cat_order, ordered=TRUE)) %>%count(category) %>%mutate(p=n / sum(n)) %>%ggplot(mapping=aes(x=category, y=n, fill=category)) +geom_col() +geom_text(mapping=aes(label=scales::percent_format(accuracy=0.1)(p)), vjust=-0.5) +scale_fill_manual(values=cat_colors) +guides(fill="none") +labs(x="", y="Species")