1、CADD药物设计
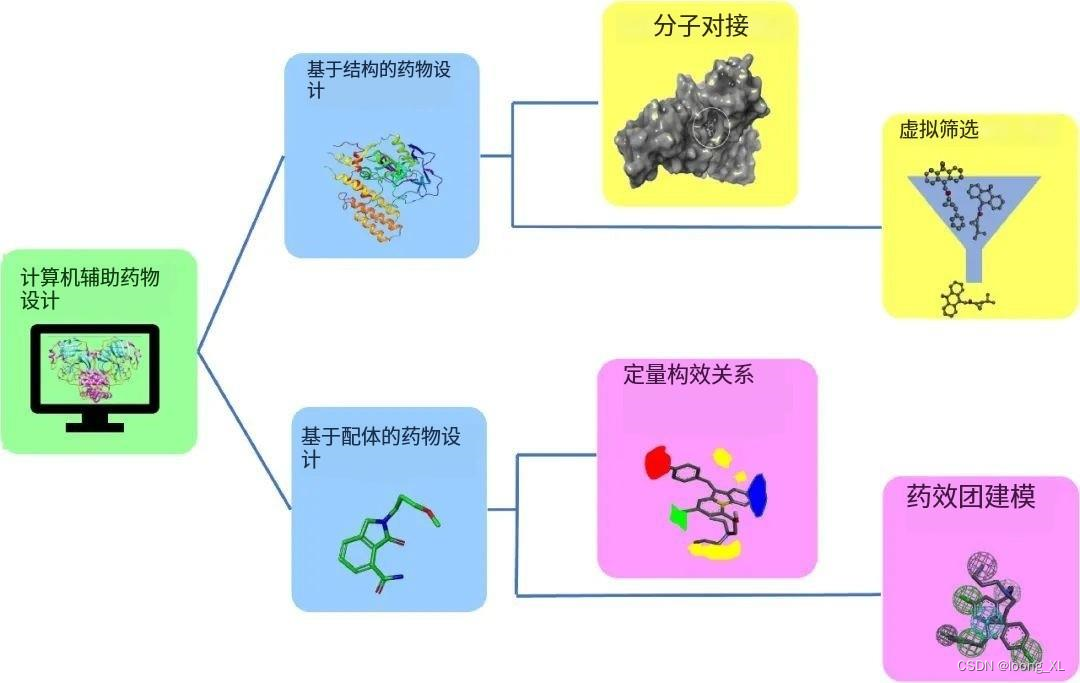
计算药物设计(CADD)是一个使用计算技术来帮助设计和开发新药的领域。它涉及使用计算机程序来模拟潜在药物分子与体内靶蛋白之间的相互作用,以及预测这些分子的性质和行为。这可以帮助研究人员识别新的药物候选物,优化其结构和活性,并预测潜在的副作用。CADD还可用于研究现有药物,了解其作用机制。它是一种强大的工具,可以帮助加速药物发现和开发过程,并已应用于包括癌症、心血管疾病和传染病在内的广泛治疗领域。
计算药物设计(CADD)涉及使用多种方法来帮助设计和开发新药。具体来说,这些方法包括:
1、计算分子对接:通过模拟药物分子与靶蛋白之间的相互作用,以评估药物的活性和可能的副作用。
2、计算分子动力学:通过模拟药物分子在体内运动和相互作用的过程,以了解其在生物体内的行为。
3、分子模拟:通过模拟药物分子的结构和性质,以预测其在体内的行为。
4、分子设计:通过对药物分子进行修饰和优化,以改善其活性和降低副作用。
5、分子结构预测:通过使用计算机模型来预测药物分子的结构和性质。
6、分子虚拟筛选:通过使用计算机程序来筛选大量化合物,以确定最有可能成为有效药物的候选物。
药效团模型与qsar模型区别:
药效团模型和 QSAR:模型都是用于研究药物分子的结构与活性之间的关系的方法。但是,它们之间有一些重要的区别:
1、药效团模型是基于药效团的概念开发的,药效团是指药物分子中具有药理活性的部分。因此,药效团模型主要关注药物分子中药效团的结构和活性之间的关系。相对而言,QSAR
模型则不需要考虑药效团的概念,可以使用整个药物分子的结构信息来预测活性。2、药效团模型的构建通常是由专家手动完成的,需要依靠专业知识和经验来确定药效团的结构和活性之间的关系。相比之下,QSAR
模型的构建则可以使用机器学习方法来自动从数据中学习药物分子的结构与活性之间的关系。3、药效团模型的准确度受到药效团的选择和模型的构建方式的影响。如果药效团的选择不当或模型的构建方式不合理,则模型的准确度可能会受到影响。相对而言,QSAR
模型的准确度则取决于使用的算法和所使用的药物分子数据的质量。通常来说,使用较为复杂的算法(如深度学习算法)或使用较大的药物分子数据集,可以得到较高的模型准确度。4、药效团模型和 QSAR
模型都可以用于药物设计和预测新药的活性。但是,药效团模型更加侧重于药物设计,因为它可以帮助确定药物分子中哪些部分具有药理活性,从而引导药物设计。相对而言,QSAR
模型则更加侧重于新药的预测,因为它可以使用整个药物分子的结构信息来预测新药的活性。总的来说,药效团模型和 QSAR 模型都是药物设计和新药预测中的重要工具,它们之间的区别在于:药效团模型更加侧重于药物设计,而 QSAR
模型更加侧重于新药的预测。
药效团模型和 QSAR 模型实现方法:
药效团模型的方法包括:
1、分子导向合成(MDS)方法:分子导向合成方法是一种以药效团为导向的药物设计方法,通过调整药效团的结构来改变药物的活性。
2、药效团反演方法(EIM):药效团反演方法是一种使用药效团模型来反推新药分子结构的方法。
3、类比构建法:类比构建法是一种使用现有药物分子的结构信息来设计新药分子的方法。
QSAR 模型的方法包括:
1、分子描述符法:分子描述符法是一种使用计算机辅助的分子描述符来表示药物分子的结构信息的方法。常见的分子描述符包括
ECFP、FCFP、MACCS 等。2、分子加权导数法:分子加权导数法是一种利用药物分子的结构信息和结构导数信息来预测药物的活性的方法。
3、机器学习、深度学习方法
2、QSAR模型
QSAR(Quantitative Structure-Activity
Relationship)是一种计算药物设计(CADD)方法,用于研究药物分子的结构与活性之间的关系。通过使用QSAR分析,研究人员可以通过对大量已知活性的药物分子进行测量和分析,来预测未知药物分子的活性。
sklearn 多元线性回归代码
import numpy as np
from sklearn.linear_model import LinearRegression# 准备药物分子的结构数据和活性数据
X = np.array([[1, 2, 3], [2, 3, 4], [3, 4, 5], [4, 5, 6]])
y = np.array([0.5, 2.5, 4.5, 6.5])# 使用线性回归模型进行训练
model = LinearRegression()
model.fit(X, y)# 使用训练好的模型预测未知药物分子的活性
X_test = np.array([[5, 6, 7]])
y_pred = model.predict(X_test)
print(y_pred) # 输出预测的活性值pytorch 多层感知机
import torch
import torch.nn as nn# 定义多层感知机模型
class MLP(nn.Module):def __init__(self, input_size, hidden_size, output_size):super(MLP, self).__init__()self.fc1 = nn.Linear(input_size, hidden_size)self.fc2 = nn.Linear(hidden_size, output_size)def forward(self, x):x = self.fc1(x)x = torch.relu(x)x = self.fc2(x)return x# 准备药物分子的结构数据和活性数据
X = torch.tensor([[1, 2, 3], [2, 3, 4], [3, 4, 5], [4, 5, 6]])
y = torch.tensor([0.5, 2.5, 4.5, 6.5])# 创建多层感知机模型
model = MLP(input_size=3, hidden_size=4, output_size=1)# 定义损失函数和优化器
criterion = nn.MSELoss()
optimizer = torch.optim.SGD(model.parameters(), lr=0.01)# 训练模型
for epoch in range(100):# 前向传播y_pred = model(X)# 计算损失loss = criterion(y_pred, y)print(f'Epoch: {epoch+1}, Loss: {loss.item():.4f}')# 清空梯度optimizer.zero_grad()# 反向传播loss.backward()# 更新参数optimizer.step()# 使用训练好的模型预测未知药物分子的活性
X_test = torch.tensor([[5, 6, 7]])
y_pred = model(X_test)
print(y_pred) # 输出预测的活性值rdkit 读取指纹 2D-SAR 例子
import pandas as pd
from rdkit import Chem
from rdkit.Chem import AllChem
from sklearn.ensemble import RandomForestRegressor# 读取药物分子的二维结构数据和活性数据
df = pd.read_csv('data.csv')
X = [Chem.MolFromSmiles(smiles) for smiles in df['SMILES']]
y = df['Activity'].values# 使用 ECFP4 描述符提取药物分子的分子描述符
X_ecfp = [AllChem.GetMorganFingerprintAsBitVect(mol, 2) for mol in X]# 使用随机森林回归模型进行训练
model = RandomForestRegressor()
model.fit(X_ecfp, y)# 使用训练好的模型预测未知药物分子的活性
X_test = [Chem.MolFromSmiles('CCCCCCCCCCCCCCCCCCCCCCCCCCCCCC')]
X_test_ecfp = [AllChem.GetMorganFingerprintAsBitVect(mol, 2) for mol in X_test]
y_pred = model.predict(X_test_ecfp)
print(y_pred) # 输出预测的活性值