<~生~信~交~流~与~合~作~请~关~注~公~众~号@生信探索>
写的数据预处理snakemake流程其实包括在每个单独的分析中比如种系遗传变异和肿瘤变异流程中,这里单独拿出来做演示用,因为数据预处理是通用的,在call变异之前需要处理好数据。
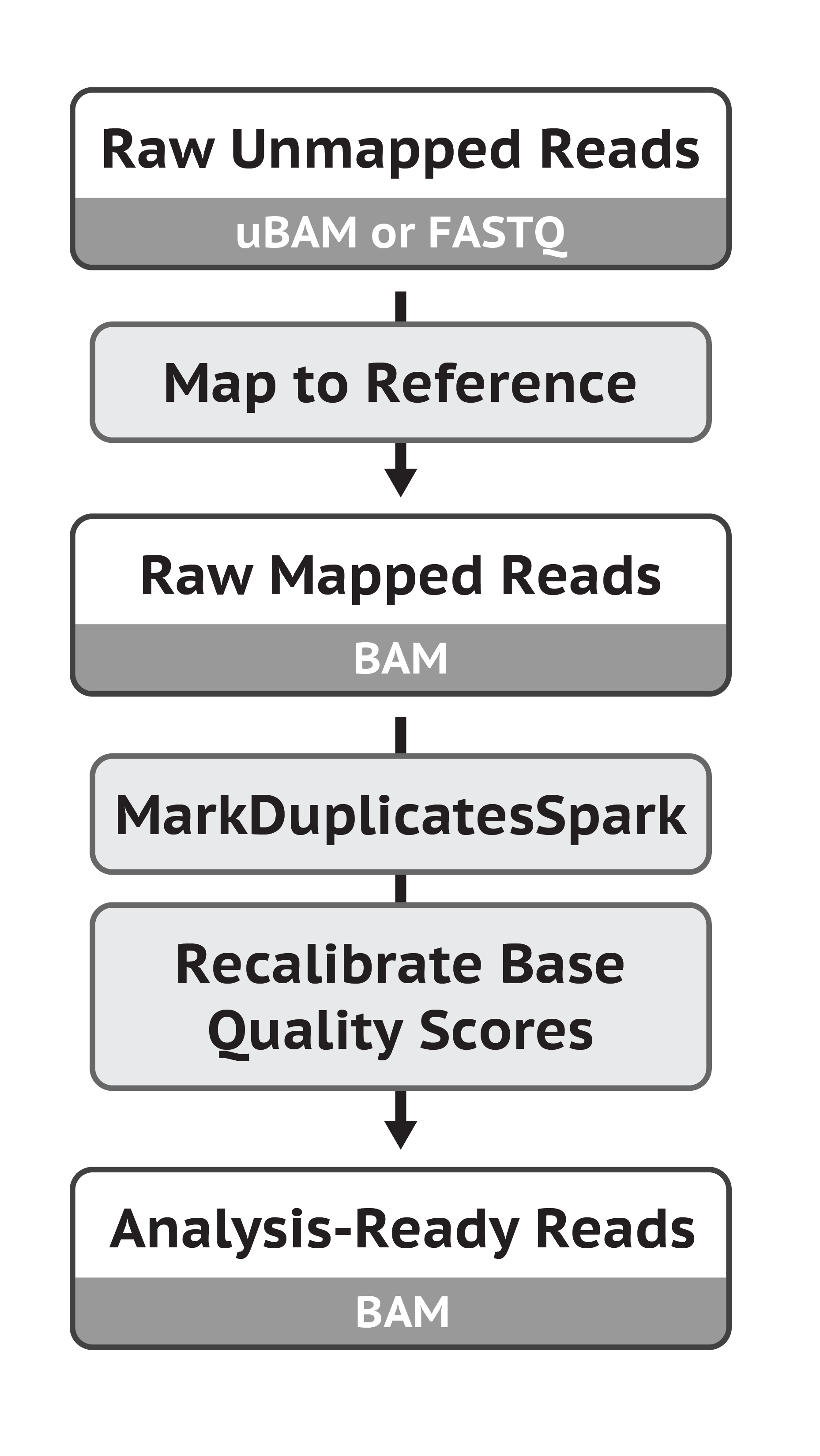
数据预处理过程包括,从fastq文件去接头、比对到基因组、去除重复、碱基质量校正,最后得到处理好的BAM或CRAM文件。
fastq去接头
fastq产生的报告json可以用multiqc汇总成一份报告
if config["fastq"].get("pe"):
rule fastp_pe:
input:
sample=get_fastq
output:
trimmed=[temp("results/trimmed/{s}{u}.1.fastq.gz"), temp("results/trimmed/{s}{u}.2.fastq.gz")],
html=temp("report/{s}{u}.fastp.html"),
json=temp("report/{s}{u}.fastp.json"),
log:
"logs/trim/{s}{u}.log"
threads: 32
wrapper:
config["warpper_mirror"]+"bio/fastp"
else:
rule fastp_se:
input:
sample=get_fastq
output:
trimmed=temp("results/trimmed/{s}{u}.fastq.gz"),
html=temp("report/{s}{u}.fastp.html"),
json=temp("report/{s}{u}.fastp.json"),
log:
"logs/trim/{s}{u}.log"
threads: 32
wrapper:
config["warpper_mirror"]+"bio/fastp"
BWA-mem2 比对+去重+排序
mem2的速度更快,所以采用。
sambaster的去除重复速度比MarkDuplicat快,所以采用。
最后用picard按照coordinate对比对结果排序。
输出的格式是CRAM,不是BAM,因为CRAM压缩效率更高,所以采用。
rule bwa_mem2:
input:
reads=get_trimmed_fastq,
reference=gatk_dict["ref"],
idx=multiext(gatk_dict["ref"], ".0123", ".amb", ".bwt.2bit.64", ".ann",".pac"),
output:
temp("results/prepared/{s}{u}.aligned.cram") # Output can be .cram, .bam, or .sam
log:
"logs/prepare/bwa_mem2/{s}{u}.log"
params:
bwa="bwa-mem2", # Can be 'bwa-mem, bwa-mem2 or bwa-meme.
extra=get_read_group,
sort="picard",
sort_order="coordinate",
dedup=config['fastq'].get('duplicates',"remove"), # Can be 'mark' or 'remove'.
dedup_extra=get_dedup_extra(),
exceed_thread_limit=True,
embed_ref=True,
threads: 32
wrapper:
config["warpper_mirror"]+"bio/bwa-memx/mem"
碱基质量校正
GATK说碱基的质量分数对call变异很重要,所以需要校正。
BaseRecalibrator计算怎么校正,ApplyBQSR更具BaseRecalibrator结果去校正。
rule BaseRecalibrator:
input:
bam="results/prepared/{s}{u}.aligned.cram",
ref=gatk_dict["ref"],
dict=gatk_dict["dict"],
known=gatk_dict["dbsnp"], # optional known sites - single or a list
output:
recal_table=temp("results/prepared/{s}{u}.grp")
log:
"logs/prepare/BaseRecalibrator/{s}{u}.log"
resources:
mem_mb=1024
params:
# extra=get_intervals(), # optional
wrapper:
config["warpper_mirror"]+"bio/gatk/baserecalibrator"
rule ApplyBQSR:
input:
bam="results/prepared/{s}{u}.aligned.cram",
ref=gatk_dict["ref"],
dict=gatk_dict["dict"],
recal_table="results/prepared/{s}{u}.grp",
output:
bam="results/prepared/{s}{u}.cram"
log:
"logs/prepare/ApplyBQSR/{s}{u}.log"
params:
embed_ref=True # embed the reference in cram output
resources:
mem_mb=2048
wrapper:
config["warpper_mirror"]+"bio/gatk/applybqsr"
索引
最后对CRAM构建所以,之后可能用得到。
rule samtools_index:
input:
"results/prepared/{s}{u}.cram"
output:
"results/prepared/{s}{u}.cram.crai"
log:
"logs/prepare/samtools_index/{s}{u}.log"
threads: 32
params:
extra="-c"
wrapper:
config["warpper_mirror"]+"bio/samtools/index"
Reference
https://gatk.broadinstitute.org/hc/en-us/articles/360035535912-Data-pre-processing-for-variant-discovery
本文由 mdnice 多平台发布