github:地址
文章目录
- Rdkit|SMARTS语言应用
- 子结构搜索HasSubstructMatch(patt)
- 得到子结构序列`GetSubstructMatch`
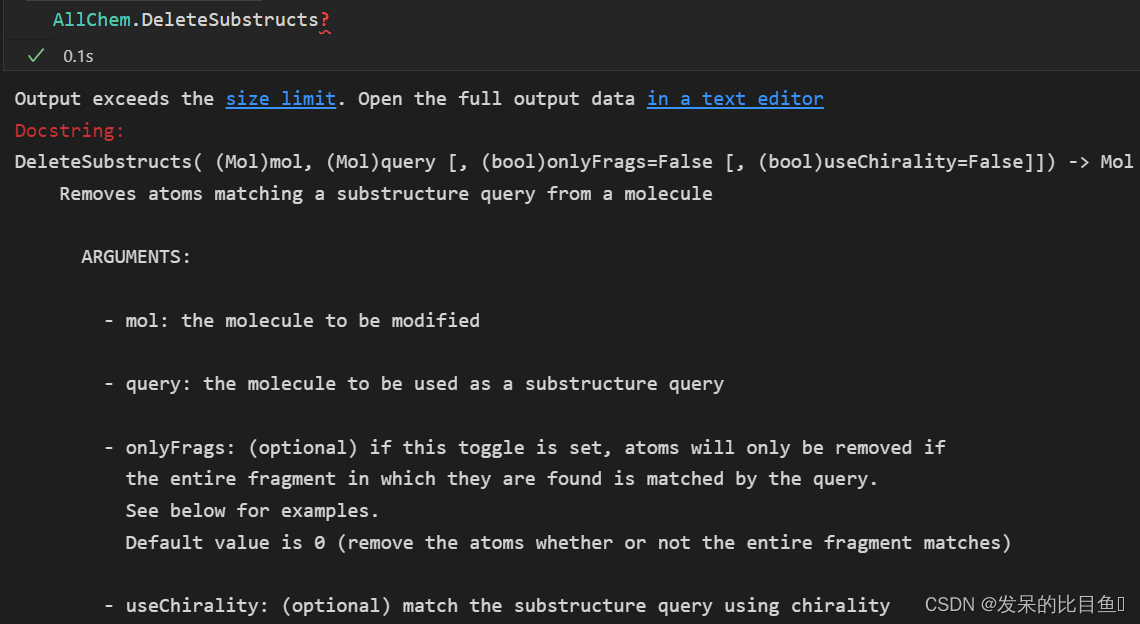
- 删除子结构`AllChem.DeleteSubstructs(m,patt)`
- 取代基替换`AllChem.ReplaceSubstructs(m, patt, rep)`
- 显示核心结构
- 显示侧链结构 `Chem.ReplaceCore`
- 分子拆分
- Mol本身为多个分子组成
- 通过分子键拆分骨架`Chem.FragmentOnBonds`
- Murcho分解`MurckoScaffold.GetScaffoldForMol`
- 參考
Rdkit|SMARTS语言应用
from rdkit import Chem
from rdkit.Chem import AllChem
from rdkit.Chem import Draw
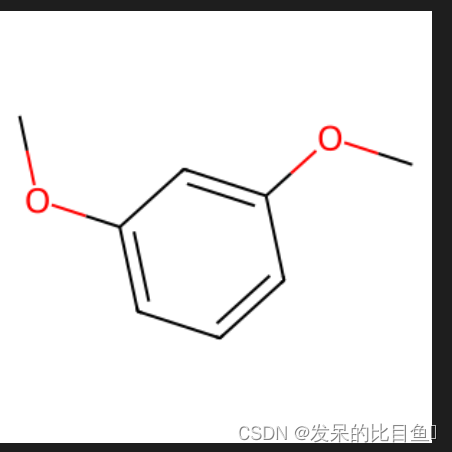
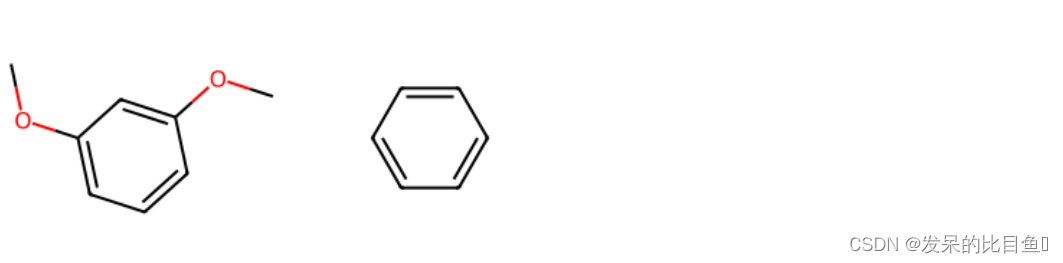
from rdkit.Chem.Scaffolds import MurckoScaffoldmol = Chem.MolFromSmiles("c1ccc(OC)cc1OC")
Chem.Draw.MolToImage(mol, size=(300,300))
子结构搜索HasSubstructMatch(patt)
- quer:需要检索的片段
- recursionPossible: (可选)
- useChirality: 能够在匹配中使用立体化学
- useQueryQueryMatches: 使用查询-查询匹配逻辑
patt = Chem.MolFromSmarts("OC") # # 导入甲氧基子结构
flag = mol.HasSubstructMatch(patt)
if flag:print("molecu m contains group -OCH3")
else:print("molecu m don't contain group -OCH3")
得到子结构序列GetSubstructMatch
query: 需要查询的分子useChirality: 能够在匹配中使用立体化学useQueryQueryMatches: 使用查询-查询匹配逻辑
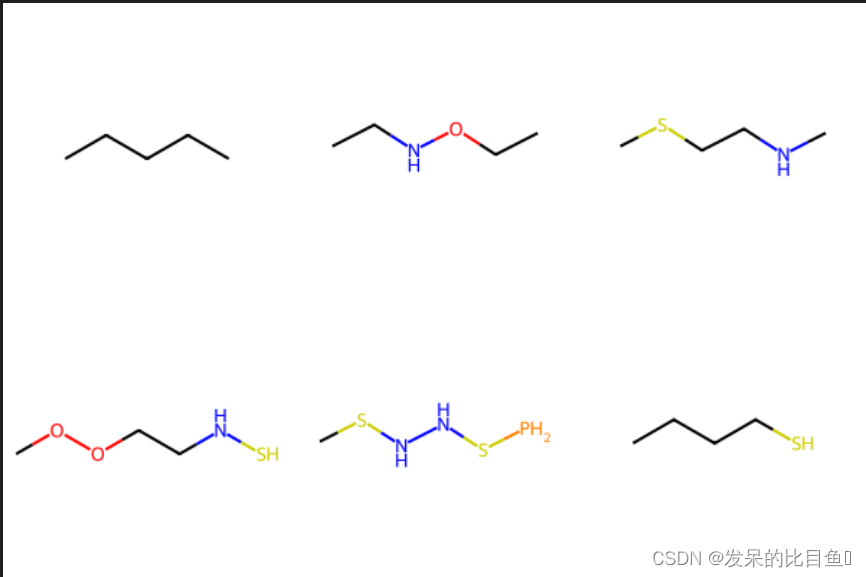
smiles_list= ['CCCCC', 'CCNOCC', 'CSCCNC', 'COOCCNS', 'CSNNSP', 'CCCCS']
mol_list = [Chem.MolFromSmiles(x) for x in smiles_list]
Chem.Draw.MolsToGridImage(mol_list)
## 匹配特定模式的分子
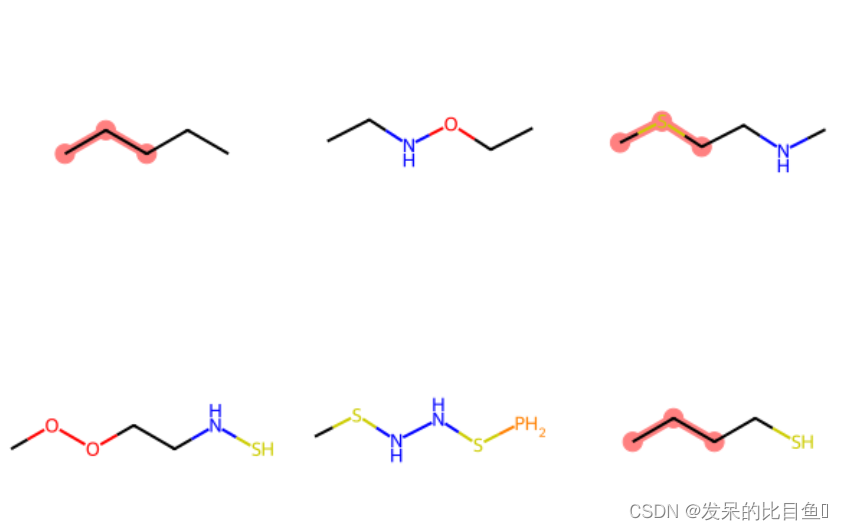
query = Chem.MolFromSmarts('C*C')
match_list = [mol.GetSubstructMatch(query) for mol in mol_list]
print(match_list)
Chem.Draw.MolsToGridImage(mols=mol_list, highlightAtomLists=match_list, molsPerRow=3)
删除子结构AllChem.DeleteSubstructs(m,patt)
- mol: 需要修饰的分子
- query: 要用作子结构查询的分子
- onlyFrags: (可选)如果设置了此切换,则只有在查询匹配原子所在的整个片段时,才会删除原子。默认值为0(删除原子,无论整个片段是否匹配)
- useChirality:(可选)使用手性匹配子结构查询
del_mol = AllChem.DeleteSubstructs(mol, patt)
mols = [mol, del_mol]
Draw.MolsToGridImage(mols, molsPerRow=4, subImgSize=(200,200),legends=['' for x in mols])
取代基替换AllChem.ReplaceSubstructs(m, patt, rep)
mol: 待修饰的分子query: 要用作子结构查询的分子replacement: 用作替代物的分子replaceAll: (可选)如果设置了此切换,则所有子结构匹配,查询将被替换为单个结果,否则每个结果都将包含单独的替代品。replacementConnectionPoint: (可选)应该与之建立键的置换中原子的索引。useChirality: (可选)使用手性匹配子结构查询
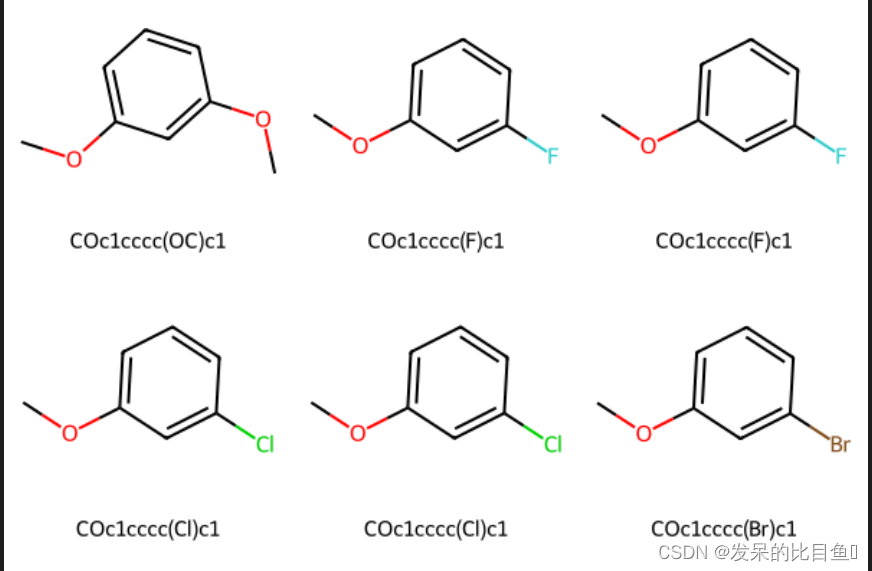
patt = Chem.MolFromSmarts("OC")
repsmis = ['F','Cl','Br','O']
mols = [mol]for r in repsmis:r_mol = Chem.MolFromSmarts(r)res = AllChem.ReplaceSubstructs(mol, patt, r_mol)mols.extend(res)smis = [Chem.MolToSmiles(mol) for mol in mols]
mols = [Chem.MolFromSmiles(smi) for smi in smis]
Draw.MolsToGridImage(mols, molsPerRow=3, subImgSize=(200,200), legends=[x for x in smis])
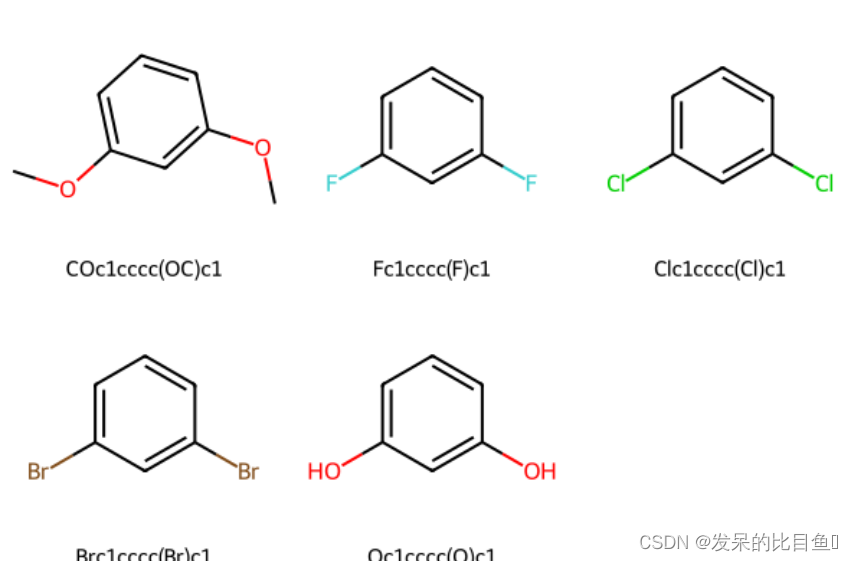
patt = Chem.MolFromSmarts("OC")
repsmis = ['F','Cl','Br','O']
mols = [mol]for r in repsmis:r_mol = Chem.MolFromSmarts(r)res = AllChem.ReplaceSubstructs(mol, patt, r_mol, replaceAll=1)mols.extend(res)smis = [Chem.MolToSmiles(mol) for mol in mols]
mols = [Chem.MolFromSmiles(smi) for smi in smis]
Draw.MolsToGridImage(mols, molsPerRow=3, subImgSize=(200,200), legends=[x for x in smis])
显示核心结构
Chem.ReplaceSidechains(m1,core) : 我们需要定义分子对象,骨架分子; 然后执行ReplaceSidechains函数,删除侧链就能得到骨架可视化。
mol: 需要修饰的分子coreQuery: 用作识别核心的子结构查询的分子useChirality: (可选)使用手性匹配子结构查询
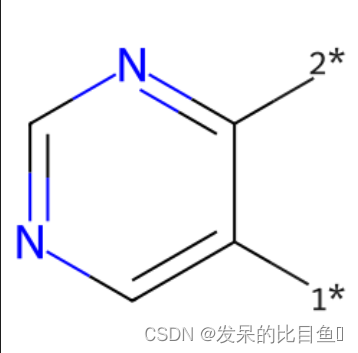
mol = Chem.MolFromSmiles('BrCCc1cncnc1C(=O)O')
core = Chem.MolFromSmiles('c1cncnc1')
tmp = Chem.ReplaceSidechains(mol,core)
Draw.MolToImage(tmp, size=(250,250))
显示侧链结构 Chem.ReplaceCore
mol: 需要修饰的分子coreQuery: 用作识别核心的子结构查询的分子matches: mol.GetSubstractMatch(…)返回的类型的匹配向量replaceDummies: 切换替换与查询中的虚设匹配的原子labelByIndex: 切换使用连接到的核心原子的索引标记连接点伪原子。requireDummyMatch: 如果分子的侧链连接在没有用假人标记的点上,它将被拒绝(返回“None”)
m1 = Chem.MolFromSmiles('BrCCc1cncnc1C(=O)O')
core = Chem.MolFromSmiles('c1cncnc1')
tmp = Chem.ReplaceCore(m1, core, labelByIndex=True)
print(tmp)
print(Chem.MolToSmiles(tmp))
Draw.MolToImage(tmp,size=(250,250))
分子拆分
Mol本身为多个分子组成
mol: 要使用的分子asMols: 可选)如果提供了这一点并且为真,则碎片将作为分子而不是原子id返回。sanitizeFrags: (可选)如果提供了这一点并且是真的,则碎片分子将在返回之前进行净化。frags:(可选,默认为None)如果asMols为true,并且这是以空列表的形式提供的,则返回时结果将为mol.GetNumAtoms()长,并且将包含每个Atom的片段分配fragsMolAtomMapping: (可选,默认为None)如果asMols为true,并且这是以空列表的形式提供的,则返回时的结果将是numFrags long,并且每个条目都将包含该片段中原子的索引:[(0,1,2,3),(4,5)]

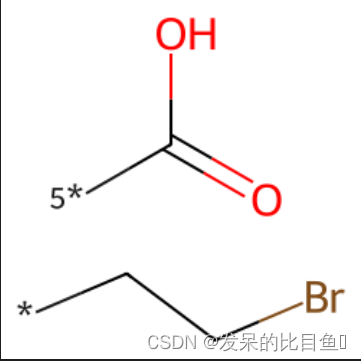
rs = Chem.GetMolFrags(tmp, asMols=True)
print(len(rs)) # 2
smi0 = Chem.MolToSmiles(rs[0])
print(smi0) # *CCBr
smi1 = Chem.MolToSmiles(rs[1])
print(smi1) # [5*]C(=O)O
通过分子键拆分骨架Chem.FragmentOnBonds
mol:需要修饰的分子bondIndices: 需要破坏键的索引addDummies:切换虚设原子的添加以指示键断裂的位置dummyLabels:用于提供用于假人的标签。每对中的第一个元素是用于替换键的beginAtom的伪元素的标签,第二个元素是用来替换键的endAtom的伪对象的标签。如果未提供,则用原子索引标记假人。bondTypes: 用于提供在片段和虚设原子之间使用的键类型。如果未提供,则默认为单个。cutsPerAtom: 用于返回在每个原子上进行的切割次数。

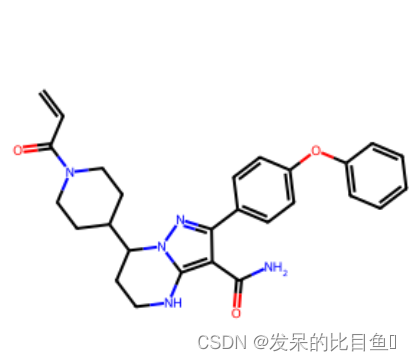
smi='C=CC(=O)N1CCC(CC1)C2CCNC3=C(C(=NN23)C4=CC=C(C=C4)OC5=CC=CC=C5)C(=O)N'
m = Chem.MolFromSmiles(smi)
Draw.MolToImage(m)
submols = m.GetSubstructMatches(Chem.MolFromSmarts('[!R][R]')) # R表示环上的原子,即匹配环上和非环上的原子
bonds_id = [m.GetBondBetweenAtoms(x,y).GetIdx() for x, y in submols] # 找到这两个原子之间的键
frags = Chem.FragmentOnBonds(m,bonds_id) # 切割得到碎片
smis= Chem.MolToSmiles(frags)
smis = smis.split('.')
mols=[m]
for smi in smis:mols.append(Chem.MolFromSmiles(smi))
Draw.MolsToGridImage(mols, molsPerRow=3, subImgSize=(300,300),legends=['' for x in mols], useSVG=True)
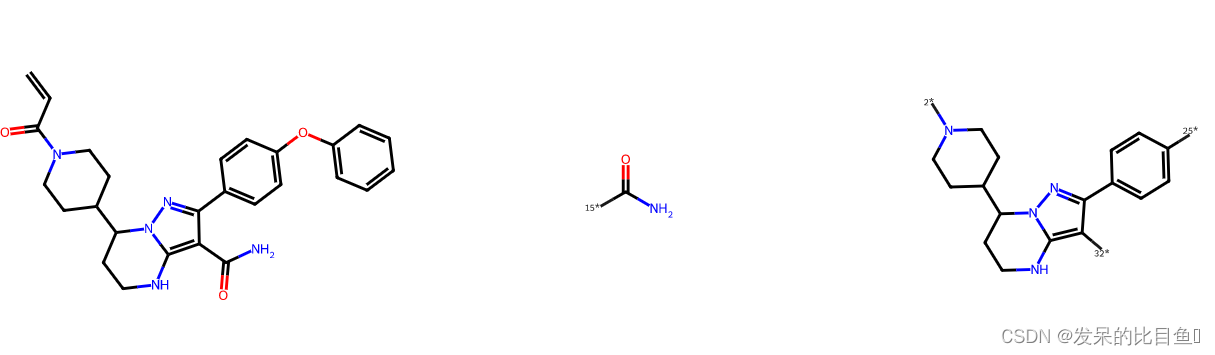
Murcho分解MurckoScaffold.GetScaffoldForMol
把分子中环结构提取出来,然后保留连接环结构的最少的键,如果该结构上的原子是双键连接,则保留双键,得到的结构称为Murcho骨架
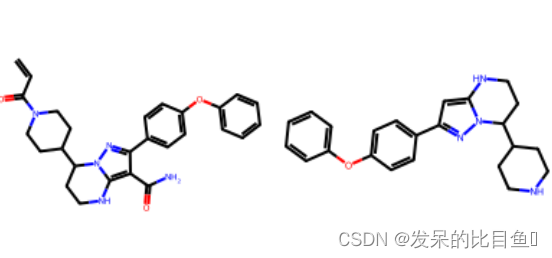
m = Chem.MolFromSmiles('C=CC(=O)N1CCC(CC1)C2CCNC3=C(C(=NN23)C4=CC=C(C=C4)OC5=CC=CC=C5)C(=O)N')
tmp = MurckoScaffold.GetScaffoldForMol(m)
Draw.MolsToImage([m, tmp])
參考
https://blog.csdn.net/qq_36801966/article/details/106564664