前言
上期我们介绍了基于 limma 来做差异表达基因,那么这期来讲一下 DESeq2,那么这两款软件有什么区别吗?区别主要在于一个是计算芯片探针给出来的结果,而 DESeq2 是基于NGS 测序结果中 Read counts 来计算差异表达,根据输入数据的不同,我们对比一下做法。
在比较高通量测序分析中,一项基本任务是分析计数数据,如 RNA-seq 中每个基因的 Read count,以获得跨实验条件的系统性变化的证据。离散性,大动态范围和异常值的存在需要一个合适的统计方法。DESeq2 是一种计数数据的差分分析方法,使用离散度和折叠变化的收缩估计来提高估计的稳定性和可解释性。这使得更多的定量分析集中在强度上,而不仅仅是差异表达的存在。下面我们就根据这篇文章的数据模式进行差异分析。

01. 软件包安装
安装 DESeq2 软件包,这个包需要通过 BiocManager 来安装,所以首先检测是否安装 BiocManager ,我之前安装过 DESeq2 ,所以不需要重复安装,如果使用 RStudio 安装不成功,可以通过 R 软件安装,运行如下:
if (!require("BiocManager", quietly = TRUE))install.packages("BiocManager")if (!require("DESeq2", quietly = TRUE))BiocManager::install("DESeq2")if (!requireNamespace("TCGAbiolinks", quietly=TRUE))BiocManager::install("TCGAbiolinks")if (!requireNamespace("EDASeq", quietly = TRUE))BiocManager::install("EDASeq")if (!requireNamespace("SummarizedExperiment", quietly = TRUE))BiocManager::install("SummarizedExperiment") if (!requireNamespace("EnhancedVolcano", quietly = TRUE))BiocManager::install("EnhancedVolcano") if (!requireNamespace("limma", quietly = TRUE))BiocManager::install("limma") library(DESeq2)
library(TCGAbiolinks)
library(EDASeq)
library(SummarizedExperiment)
library(EnhancedVolcano)
library(limma)02. TCGA 数据读取
这次我们选择 TCGA 数据的RNA-SEQ的 Reads count 数据,一般后缀为 HTSeq-counts.txt。
我们看通过 TCGAbiolinks 这个软件包都可以获得哪些数据库的数据集,TCGA 全部数据集,还是非常全面的,如下:
getGDCprojects()$project_id## [1] "TCGA-BRCA" "GENIE-MSK" "GENIE-VICC" "GENIE-UHN"
## [5] "CPTAC-2" "CMI-ASC" "BEATAML1.0-COHORT" "CGCI-BLGSP"
## [9] "BEATAML1.0-CRENOLANIB" "CMI-MPC" "CMI-MBC" "GENIE-GRCC"
## [13] "GENIE-MDA" "GENIE-JHU" "GENIE-NKI" "FM-AD"
## [17] "VAREPOP-APOLLO" "WCDT-MCRPC" "GENIE-DFCI" "TARGET-ALL-P3"
## [21] "TARGET-ALL-P2" "OHSU-CNL" "TARGET-ALL-P1" "MMRF-COMMPASS"
## [25] "TARGET-CCSK" "ORGANOID-PANCREATIC" "NCICCR-DLBCL" "TARGET-NBL"
## [29] "TARGET-OS" "TARGET-RT" "TARGET-WT" "TCGA-LAML"
## [33] "CGCI-HTMCP-CC" "TARGET-AML" "HCMI-CMDC" "TCGA-DLBC"
## [37] "TCGA-CHOL" "CTSP-DLBCL1" "TRIO-CRU" "TCGA-MESO"
## [41] "TCGA-ACC" "TCGA-UCS" "TCGA-KICH" "TCGA-PCPG"
## [45] "TCGA-ESCA" "TCGA-THYM" "TCGA-TGCT" "TCGA-UVM"
## [49] "TCGA-CESC" "TCGA-BLCA" "TCGA-PAAD" "TCGA-LIHC"
## [53] "TCGA-SKCM" "TCGA-UCEC" "TCGA-PRAD" "REBC-THYR"
## [57] "TCGA-THCA" "TCGA-OV" "TCGA-LGG" "TCGA-SARC"
## [61] "CPTAC-3" "TCGA-COAD" "TCGA-READ" "TCGA-KIRP"
## [65] "TCGA-GBM" "TCGA-STAD" "TCGA-LUAD" "TCGA-KIRC"
## [69] "TCGA-LUSC" "TCGA-HNSC"使用TCGAbiolinks:::getProjectSummary(project)查看project中有哪些数据类型,如查询"TCGA-COAD",有8种数据类型,case_count为病人数,file_count为对应的文件数。要下载表达谱,可以设置data.category=“Transcriptome Profiling”,如下:
TCGAbiolinks:::getProjectSummary("TCGA-COAD")## $file_count
## [1] 15701
##
## $data_categories
## file_count case_count data_category
## 1 2971 460 Copy Number Variation
## 2 531 461 Clinical
## 3 2835 461 Biospecimen
## 4 2493 459 Transcriptome Profiling
## 5 3952 433 Simple Nucleotide Variation
## 6 363 360 Proteome Profiling
## 7 556 458 DNA Methylation
## 8 2000 460 Sequencing Reads
##
## $case_count
## [1] 461
##
## $file_size
## [1] 2.747227e+13现在我们选择一个结肠癌的表达数据,比较癌和癌旁组织之间的表达差异基因,下载 TCGA-COAD,下载方式可以选择直接下载:https://portal.gdc.cancer.gov/{.uri} 下载 HTSeq-counts.txt 和临床数据,也可以通过 TCGAbiolinks 软件包下载。
# 请求数据。
query <- GDCquery(project ="TCGA-COAD",data.category = "Transcriptome Profiling",data.type ="Gene Expression Quantification" ,workflow.type="HTSeq - Counts")
getResults(query, rows, cols) 根据指定行名或列名从 query 中获取结果,此处用来获得样本的 barcode,检索出519个 barcodes。从 samplesDown 中筛选出TP(实体肿瘤)样本的barcodes,如下:
samplesDown <- getResults(query,cols=c("cases"))我们需要对数据进行处理,整理出需要的数据,这里我们只是比较癌和癌旁的表达差异,那么临床数据需要找到样本的对应分组。之前我们给出样本类型的简写,实体瘤样本为 Primary Solid Tumor 即为 TP, 正常样本为 Solid Tissue Normal 即为 NT。通过 TCGAquery_SampleTypes(barcode, typesample) 获得 478个 TP样本barcodes,41个NT样本barcodes。
dataSmTP <- TCGAquery_SampleTypes(barcode = samplesDown,typesample = "TP")
dataSmNT <- TCGAquery_SampleTypes(barcode = samplesDown,typesample = "NT")将样本与分组关联起来,以便有序过滤样本,并保证能够对应修改分组信息,如下:
group<-as.data.frame(list(Sample=c(dataSmTP,dataSmNT),Group=c(rep("TP",length(dataSmTP)),rep("NT",length(dataSmNT)))))设置 barcodes 参数,筛选符合要求的 478 个肿瘤样本数据和 41 正常组织数据,根据传入 barcodes 进行数据过滤,如下:
queryDown <- GDCquery(project = "TCGA-COAD",data.category = "Transcriptome Profiling",data.type = "Gene Expression Quantification", workflow.type = "HTSeq - Counts", barcode = c(dataSmTP, dataSmNT))
下载数据,默认存放位置为当前工作目录下的GDCdata文件夹中。
-
method ;“API"或者"client”,"API"速度更快,但是容易下载中断;
-
directory:下载文件的保存地址。Default: GDCdata;
-
files.per.chunk = NULL:使用API下载大文件的时候,可以把文件分成几个小文件来下载,可以解决下载容易中断的问题。
GDCdownload(queryDown,method = "api", directory = "GDCdata",files.per.chunk = 10)03. 数据处理
GDCprepare()将前面GDCquery()的结果准备成R语言可处理的SE(SummarizedExperiment)文件
读取下载的数据并将其准备到R对象中,在工作目录生成(save=TRUE)COAD_case.rda文件。GDCprepare():Prepare GDC data,准备GDC数据,使其可用于R语言中进行分析
dataPrep1 <- GDCprepare(query = queryDown, save = TRUE, save.filename ="COAD_case.rda")
获得的结果如下:
dataPrep1## class: RangedSummarizedExperiment
## dim: 56602 519
## metadata(1): data_release
## assays(1): HTSeq - Counts
## rownames(56602): ENSG00000000003 ENSG00000000005 ... ENSG00000281912 ENSG00000281920
## rowData names(3): ensembl_gene_id external_gene_name original_ensembl_gene_id
## colnames(519): TCGA-D5-6530-01A-11R-1723-07 TCGA-G4-6320-01A-11R-1723-07 ...
## TCGA-AA-3712-11A-01R-1723-07 TCGA-AA-3522-11A-01R-A32Z-07
## colData names(107): barcode patient ... paper_vascular_invasion_present
## paper_vital_status去除dataPrep1中的异常值,dataPrep1数据中含有肿瘤组织和正常组织的数据,生产热图,以及数据,函数功能描述:Array Array Intensity correlation (AAIC) and correlation boxplot to define outlier,如下:
dataPrep2 <- TCGAanalyze_Preprocessing(object = dataPrep1,cor.cut = 0.6,datatype = "HTSeq - Counts")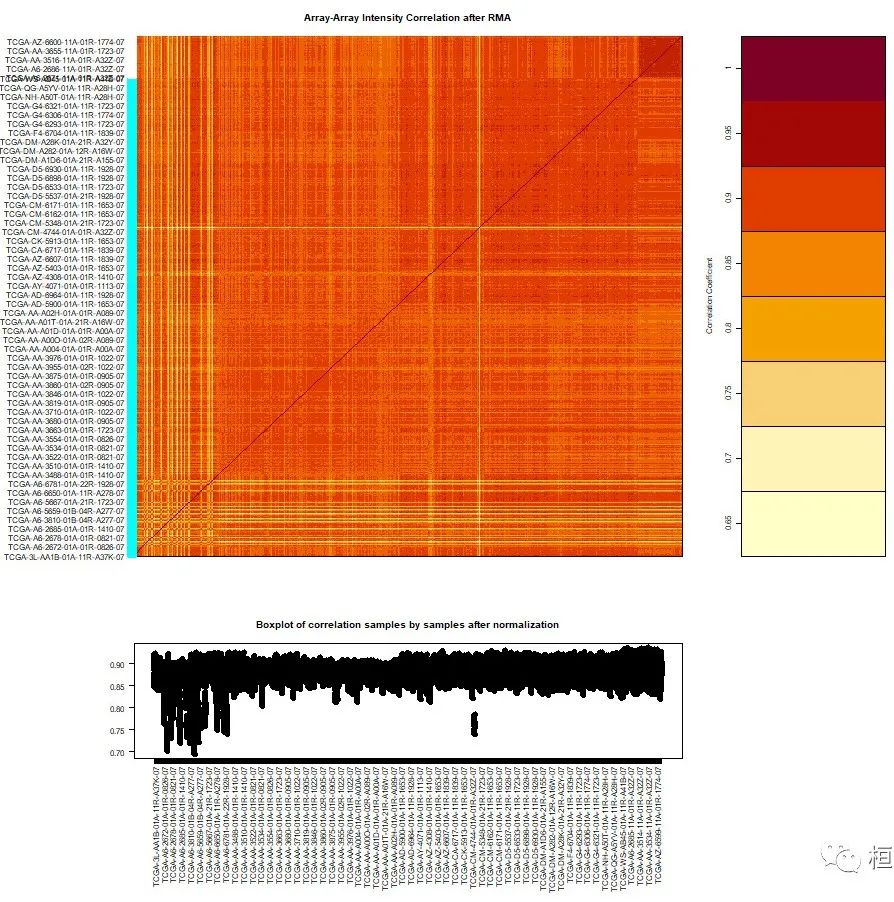
获得一个 counts 矩阵, 如下:
dim(dataPrep2)## [1] 56602 519
dataPrep2[1:5,1:3]## TCGA-3L-AA1B-01A-11R-A37K-07 TCGA-4N-A93T-01A-11R-A37K-07
## ENSG00000000003 7280 7164
## ENSG00000000005 23 67
## ENSG00000000419 2065 2632
## ENSG00000000457 869 916
## ENSG00000000460 466 266
## TCGA-4T-AA8H-01A-11R-A41B-07
## ENSG00000000003 2927
## ENSG00000000005 89
## ENSG00000000419 848
## ENSG00000000457 370
## ENSG00000000460 214```
将预处理后的数据dataPrep2,写入新文件"COAD_dataPrep.csv"
write.csv(dataPrep2,file = "COAD_dataPrep.csv",quote = FALSE)TCGAtumor_purity(barcodes, estimate, absolute, lump, ihc, cpe),使用来自5种方法的5个估计值作为阈值对 TCGA 样本进行过滤,这5个值是estimate, absolute, lump, ihc, cpe,这里设置cpe=0.6(cpe是派生的共识度量,是将所有方法的标准含量归一化后的均值纯度水平,以使它们具有相等的均值和标准差)。筛选肿瘤纯度大于等于60%的样本数据,如下:
purityDATA <- TCGAtumor_purity(colnames(dataPrep1), 0, 0, 0, 0, 0.6)## the following TCGA barcodes do not have info on tumor purity:
## [1] "TCGA-A6-2672-01B-03R-2302-07" "TCGA-AZ-6601-11A-01R-1774-07" "TCGA-A6-2686-11A-01R-A32Z-07"
## [4] "TCGA-AZ-6603-11A-02R-1839-07" "TCGA-AZ-6605-11A-01R-1839-07" "TCGA-AA-3660-11A-01R-1723-07"
## [7] "TCGA-AA-3713-11A-01R-1723-07" "TCGA-AZ-6598-11A-01R-1774-07" "TCGA-A6-2680-11A-01R-A32Z-07"
## [10] "TCGA-AA-3655-11A-01R-1723-07" "TCGA-AA-3516-11A-01R-A32Z-07" "TCGA-F4-6704-11A-01R-1839-07"
## [13] "TCGA-AZ-6600-11A-01R-1774-07" "TCGA-A6-5659-11A-01R-1653-07" "TCGA-AA-3520-11A-01R-A32Z-07"
## [16] "TCGA-A6-2679-11A-01R-A32Z-07" "TCGA-A6-2678-11A-01R-A32Z-07" "TCGA-AZ-6599-11A-01R-1774-07"
## [19] "TCGA-A6-5662-11A-01R-1653-07" "TCGA-A6-2683-11A-01R-A32Z-07" "TCGA-AA-3525-11A-01R-A32Z-07"
## [22] "TCGA-AA-3527-11A-01R-A32Z-07" "TCGA-AA-3489-11A-01R-1839-07" "TCGA-A6-2685-11A-01R-A32Z-07"
## [25] "TCGA-A6-5665-11A-01R-1653-07" "TCGA-AA-3697-11A-01R-1723-07" "TCGA-AA-3496-11A-01R-1839-07"
## [28] "TCGA-AA-3534-11A-01R-A32Z-07" "TCGA-A6-2682-11A-01R-A32Z-07" "TCGA-AA-3663-11A-01R-1723-07"
## [31] "TCGA-A6-5667-11A-01R-1723-07" "TCGA-AA-3517-11A-01R-A32Z-07" "TCGA-AA-3518-11A-01R-1672-07"
## [34] "TCGA-A6-2675-11A-01R-1723-07" "TCGA-A6-2671-11A-01R-A32Z-07" "TCGA-AA-3662-11A-01R-1723-07"
## [37] "TCGA-AA-3531-11A-01R-A32Z-07" "TCGA-AA-3511-11A-01R-1839-07" "TCGA-A6-2684-11A-01R-A32Z-07"
## [40] "TCGA-AA-3514-11A-01R-A32Z-07" "TCGA-AA-3712-11A-01R-1723-07" "TCGA-AA-3522-11A-01R-A32Z-07"filtered 为被过滤的数据, pure_barcodes是我们要的肿瘤数据
Purity.COAD<-purityDATA$pure_barcodes
length(Purity.COAD)
## [1] 450
normal.COAD<-purityDATA$filtered
length(normal.COAD)
## [1] 42```
获取肿瘤纯度大于60%的450个肿瘤组织样本,42个正常组织样本,共计492个样本
puried_data <-dataPrep2[,c(Purity.COAD,normal.COAD)]
puried_data[1:5,1:3]## TCGA-D5-6530-01A-11R-1723-07 TCGA-G4-6320-01A-11R-1723-07
## ENSG00000000003 2241 5323
## ENSG00000000005 2 75
## ENSG00000000419 1181 1168
## ENSG00000000457 591 616
## ENSG00000000460 290 302
## TCGA-AD-6888-01A-11R-1928-07
## ENSG00000000003 8152
## ENSG00000000005 105
## ENSG00000000419 5375
## ENSG00000000457 771
## ENSG00000000460 644基因注释,需要加载"SummarizedExperiment"包,"SummarizedExperiment container"每个由数字或其他模式的类似矩阵的对象表示。通常表示感兴趣的基因组范围,列代表样品。
rowData(dataPrep1)
## DataFrame with 56602 rows and 3 columns
## ensembl_gene_id external_gene_name original_ensembl_gene_id
## <character> <character> <character>
## ENSG00000000003 ENSG00000000003 TSPAN6 ENSG00000000003.13
## ENSG00000000005 ENSG00000000005 TNMD ENSG00000000005.5
## ENSG00000000419 ENSG00000000419 DPM1 ENSG00000000419.11
## ENSG00000000457 ENSG00000000457 SCYL3 ENSG00000000457.12
## ENSG00000000460 ENSG00000000460 C1orf112 ENSG00000000460.15
## ... ... ... ...
## ENSG00000281904 ENSG00000281904 AC233263.6 ENSG00000281904.1
## ENSG00000281909 ENSG00000281909 HERC2P7 ENSG00000281909.1
## ENSG00000281910 ENSG00000281910 SNORA50A ENSG00000281910.1
## ENSG00000281912 ENSG00000281912 LINC01144 ENSG00000281912.1
## ENSG00000281920 ENSG00000281920 AC007389.5 ENSG00000281920.1传入数据 dataPrep1 必须为 SummarizedExperiment 对象
rownames(puried_data)<-rowData(dataPrep1)$external_gene_name将结果写入文件"puried.COAD.cancer.csv"
write.csv(puried_data,file = "puried.COAD.csv",quote = FALSE)将标准化后的数据再过滤,如下:
dataNorm <- TCGAanalyze_Normalization(tabDF = puried_data,geneInfo = geneInfo,method = "gcContent")## I Need about 348 seconds for this Complete Normalization Upper Quantile [Processing 80k elements /s]
## Step 1 of 4: newSeqExpressionSet ...
## Step 2 of 4: withinLaneNormalization ...
## Step 3 of 4: betweenLaneNormalization ...
## Step 4 of 4: .quantileNormalization ...去除掉表达量较低(count较低)的基因,得到最终的数据,如下:
dataFilt <- TCGAanalyze_Filtering(tabDF = dataNorm,method = "quantile", qnt.cut = 0.25)str(dataFilt)
## num [1:13125, 1:492] 442 0 5632 185 1326 ...
## - attr(*, "dimnames")=List of 2
## ..$ : chr [1:13125] "A1CF" "A2ML1" "A2M" "A4GALT" ...
## ..$ : chr [1:492] "TCGA-D5-6530-01A-11R-1723-07" "TCGA-G4-6320-01A-11R-1723-07" "TCGA-AD-6888-01A-11R-1928-07" "TCGA-CK-6747-01A-11R-1839-07" ...dataFilt[1:5,1:3]
## TCGA-D5-6530-01A-11R-1723-07 TCGA-G4-6320-01A-11R-1723-07 TCGA-AD-6888-01A-11R-1928-07
## A1CF 442 420 1471
## A2ML1 0 0 8
## A2M 5632 2389 1530
## A4GALT 185 102 169
## AAAS 1326 1456 1397```
04. 差异表达基因分析
在做差异表达时,输入文件要求必须是 counts 矩阵,那么我们将上面整理后得到的 dataFilt 做成矩阵,如下:
exp<-as.matrix(dataFilt)
rownames(exp)[1:100]## [1] "A1CF" "A2ML1" "A2M" "A4GALT" "AAAS" "AACS" "AADAC" "AADAT"
## [9] "AAGAB" "AAK1" "AAMP" "AARS2" "AARSD1" "AARS" "AASDHPPT" "AASDH"
## [17] "AASS" "AATF" "AATK" "ABAT" "ABCA10" "ABCA11P" "ABCA12" "ABCA13"
## [25] "ABCA17P" "ABCA1" "ABCA2" "ABCA3" "ABCA5" "ABCA6" "ABCA7" "ABCA8"
## [33] "ABCA9" "ABCB10" "ABCB1" "ABCB4" "ABCB6" "ABCB7" "ABCB8" "ABCB9"
## [41] "ABCC10" "ABCC13" "ABCC1" "ABCC2" "ABCC3" "ABCC4" "ABCC5" "ABCC6P1"
## [49] "ABCC6P2" "ABCC6" "ABCC9" "ABCD1" "ABCD3" "ABCD4" "ABCE1" "ABCF1"
## [57] "ABCF2" "ABCF3" "ABCG1" "ABCG2" "ABCG5" "ABHD10" "ABHD11" "ABHD12B"
## [65] "ABHD12" "ABHD13" "ABHD14A" "ABHD14B" "ABHD15" "ABHD2" "ABHD3" "ABHD4"
## [73] "ABHD5" "ABHD6" "ABHD8" "ABI1" "ABI2" "ABI3BP" "ABI3" "ABL1"
## [81] "ABL2" "ABLIM1" "ABLIM2" "ABLIM3" "ABO" "ABR" "ABT1" "ABTB1"
## [89] "ABTB2" "ACAA1" "ACAA2" "ACACA" "ACACB" "ACAD10" "ACAD8" "ACAD9"
## [97] "ACADM" "ACADSB" "ACADS" "ACADVL"数据整理并且过滤后,此时获得行为 13125 个基因,列为 492 个样本的基因表达矩阵,如下:
dim(exp)
## [1] 13125 492
对表达矩阵的相同行取平均值,利用 limma 包中函数 avereps 进行计算,其实在这些做差异分析的数据包有些也可以兼容使用,哪个方法方便实用我们就选择哪个函数,这个都无所谓,最后计算的结果,如下:
data=avereps(exp)
dim(data)
## [1] 13125 492DEGSeq2 这个包要求表达值必须为整数,所以我们需要把矩阵中的数值进行取整数,利用 round 函数,如下
data=round(data,0)设计分组信息,起初我们根据 TP 和 NT 样本信息共检索到519个样本,由于我们上面对不符合标准的样本进行了一定的过滤,所以需要重新整理样本分组信息,如下:
head(group)
## Sample Group
## 1 TCGA-D5-6530-01A-11R-1723-07 TP
## 2 TCGA-G4-6320-01A-11R-1723-07 TP
## 3 TCGA-AD-6888-01A-11R-1928-07 TP
## 4 TCGA-CK-6747-01A-11R-1839-07 TP
## 5 TCGA-AA-3975-01A-01R-1022-07 TP
## 6 TCGA-A6-6780-01A-11R-1839-07 TPtable(group$Group)
##
## NT TP
## 41 478```
```
根据我们之前整理的临床分组,癌组织478个,正常组织41个,过滤后的样本数量临床分组的样本492,癌组织样本451,正常组织样本41个,如下:```
group1=group[group$Sample %in% colnames(exp),]
table(group1$Group)##
## NT TP
## 41 451```
DESeq2 软件包中 DESeqDataSetFromMatrix 函数要求分组设计格式,如下:
design=as.factor(group1$Group)上面一系列操作都是为了达到 DESeq2 的输入文件的标准,但其实主程序非常简单,一行命令搞定所有,如下:
dds<-DESeqDataSetFromMatrix(data,DataFrame(design),design = ~design)
dds<-DESeq(dds,fitType = "local") ## or mean
res<-as.data.frame(results(dds))
head(res)## baseMean log2FoldChange lfcSE stat pvalue padj
## A1CF 1204.033014 -1.2743230 0.16883176 -7.547887 4.423763e-14 1.361676e-13
## A2ML1 7.896115 3.5771758 0.57556789 6.215037 5.131255e-10 1.225841e-09
## A2M 11164.726955 -1.4163123 0.14667565 -9.656083 4.632373e-22 2.233648e-21
## A4GALT 379.188423 -0.7515222 0.16140586 -4.656102 3.222520e-06 5.937889e-06
## AAAS 1537.066486 0.4436685 0.05680360 7.810569 5.693031e-15 1.843143e-14
## AACS 2016.414047 0.1868876 0.07884018 2.370461 1.776591e-02 2.343257e-02```
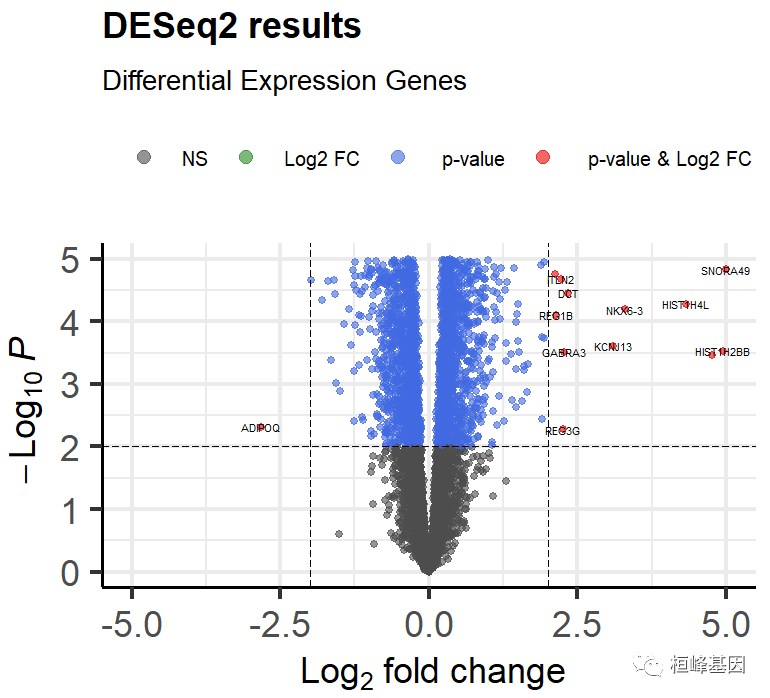
05. 绘制火山图
差异基因完成之后,我们进行火山图的绘制,这个方法比较好用我们在讲解 limma 软件包做差异分析的时候已经使用过,我们稍微改一下既可以使用,如下:
require(EnhancedVolcano)
EnhancedVolcano(res,lab = rownames(res),labSize = 2,x = "log2FoldChange",y = "pvalue",xlab = bquote(~Log[2]~ "fold change"),ylab = bquote(~-Log[10]~italic(P)),pCutoff = 0.01,## pvalue闃堝€?FCcutoff = 2,## FC cutoffxlim = c(-5,5),ylim = c(0,5),colAlpha = 0.6,legendLabels =c("NS","Log2 FC"," p-value"," p-value & Log2 FC"),legendPosition = "top",legendLabSize = 10,legendIconSize = 3.0,pointSize = 1.5,title ="DESeq2 results",subtitle = 'Differential Expression Genes',)
我们在做整套分析时需要注意使用硬件的配置,我是在window 11 利用 Rstudio 做的分析,可想而知,还是很耗内存的,看下我电脑的配置,如下:
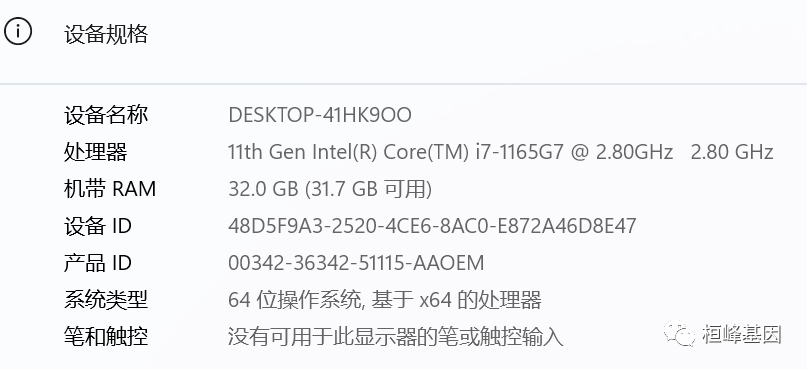
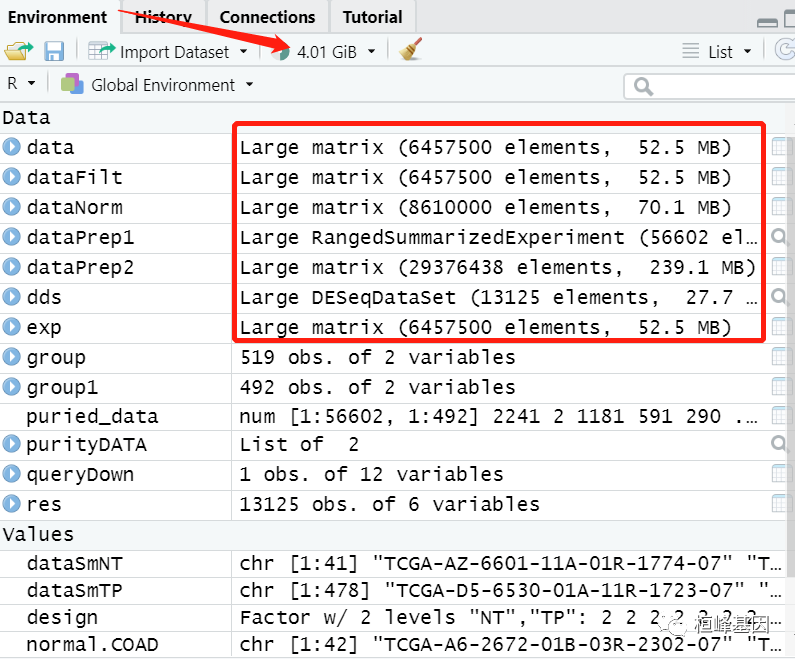
然后我们再看一下做这几百个样本需要的内存,当然这中间产生了很多切换变量,如果可以优化的后期我会尽量优化,如下:
关于差异表达主流软件已经都了解的差不多了,后面会针对检测出来的表达基因做一些后续的分析,比如GO, KEGG, GSEA等功能上的分析,敬请期待!
关注公众号,每日有更新!

桓峰基因
生物信息分析,SCI文章撰写及生物信息基础知识学习:R语言学习,perl基础编程,linux系统命令,Python遇见更好的你
38篇原创内容
公众号
Reference:
-
Agarwal A, Koppstein D, Rozowsky J, et al. Comparison and calibration of transcriptome data from RNA-Seq and tiling arrays. BMC Genomics. 2010;11:383.
-
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139‐140.
-
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550.
-
Maurya NS, Kushwaha S, Chawade A, Mani A. Transcriptome profiling by combined machine learning and statistical R analysis identifies TMEM236 as a potential novel diagnostic biomarker for colorectal cancer. Sci Rep. 2021;11(1):14304.