我们在使用软件包时总是不断的出现各种各样的bug,尤其对于初学者简直就是科研路上的拦路虎,死死的挡住了科研的道路,鉴于小编写公众号时同样会遇到各种问题和困难,因此特意将这些bug记录下来,并分析给大家解决办法,希望尽微薄之力助大家一臂之力。
今天就分析一个单细胞富集分析报错 (singleseqgset)的问题以及解决办法。
报错信息:
logfc.data <- logFC(cluster.ids=as.vector(ser@meta.data$real_id),expr.mat=ser@assays$RNA$data)
Error in if (!class(expr.mat) == "dgCMatrix") { : the condition has length > 1问题解读:
这个报错信息是说 if 条件的长度大于1 也就是出现多个 TRUE,或者 FALSE, 这样肯定是不对,那么问题出现在class,那么我们看下class的使用方法:
class (as.matrix (1)) "matrix" "array" This is because an array in R is a vector with multiple dimensions, and the matrix is a special type of array with exactly two dimensions, so every matrix is an array.
https://bioinformatics.stackexchange.com/questions/16362/converting-a-matrix-array-to-matrix#:~:text=%3E%20class%20%28as.matrix%20%281%29%29%20%22matrix%22%20%22array%22%20This%20is,a%20matrix%20is%20telling%20us%20here%2C%20see%20http%3A%2F%2Fadv-r.had.co.nz%2FData-structures.html
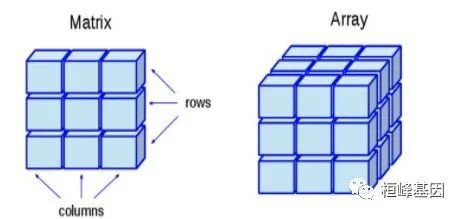
expr.mat <- matrix(c(3:14), nrow = 4, byrow = TRUE)
print(expr.mat)
# [,1] [,2] [,3]
# [1,] 3 4 5
# [2,] 6 7 8
# [3,] 9 10 11
# [4,] 12 13 14
class(expr.mat)
# [1] "matrix" "array"我们注意到执行class(expr.mat)后其实是获得两个元素的向量,因此会报错:the condition has length > 1
解决办法:
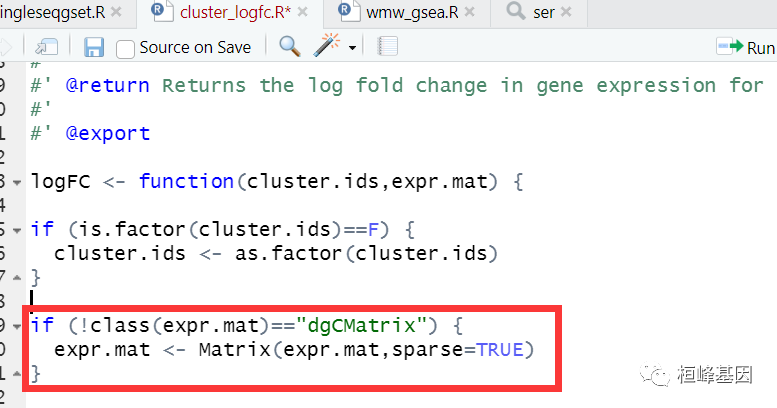
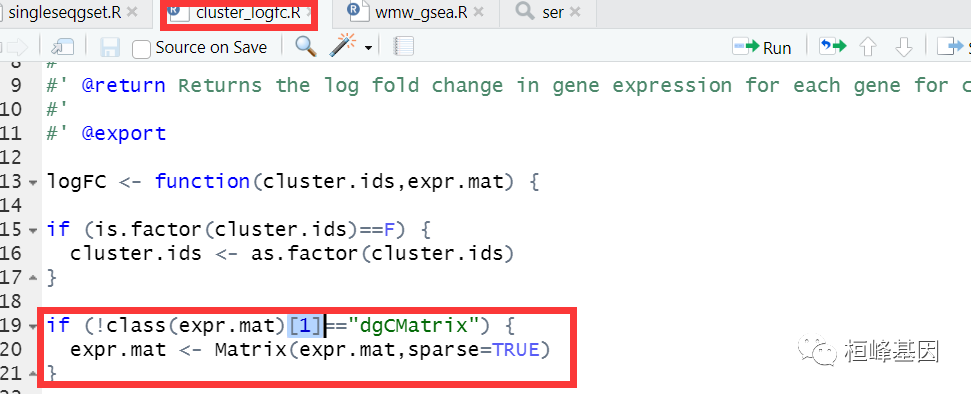
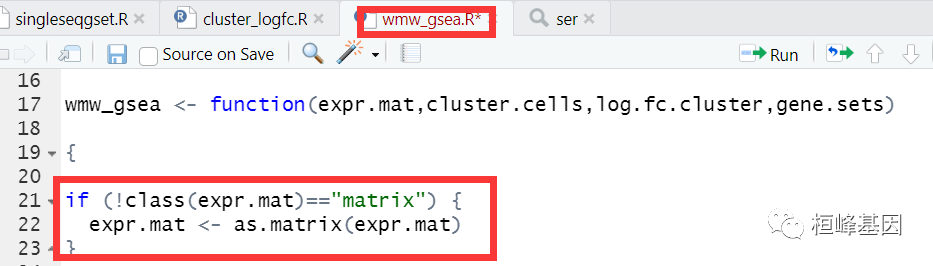
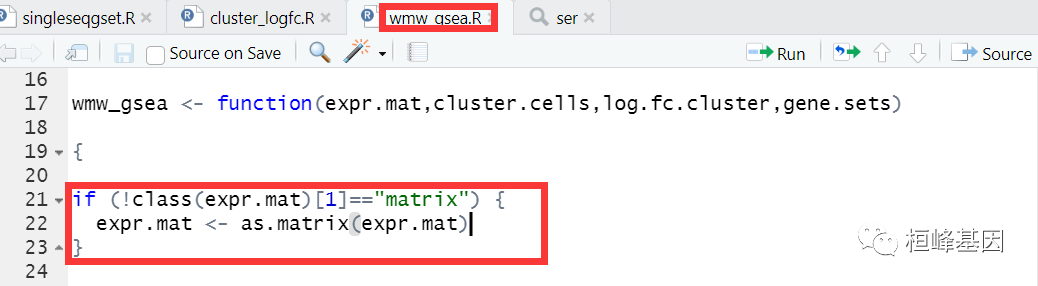
将 if (!class(expr.mat) == "dgCMatrix") 改为 if (!class(expr.mat)[1] == "dgCMatrix")
修改之后我们再运行:
if (!class(expr.mat)[1] == "dgCMatrix") {print(expr.mat)}
# [,1] [,2] [,3]
# [1,] 3 4 5
# [2,] 6 7 8
# [3,] 9 10 11
# [4,] 12 13 14这样就可以运行了,具体看下 singleseqgset 实操代码:
下载软件包:在git上找到singleseqgset下载到本地。
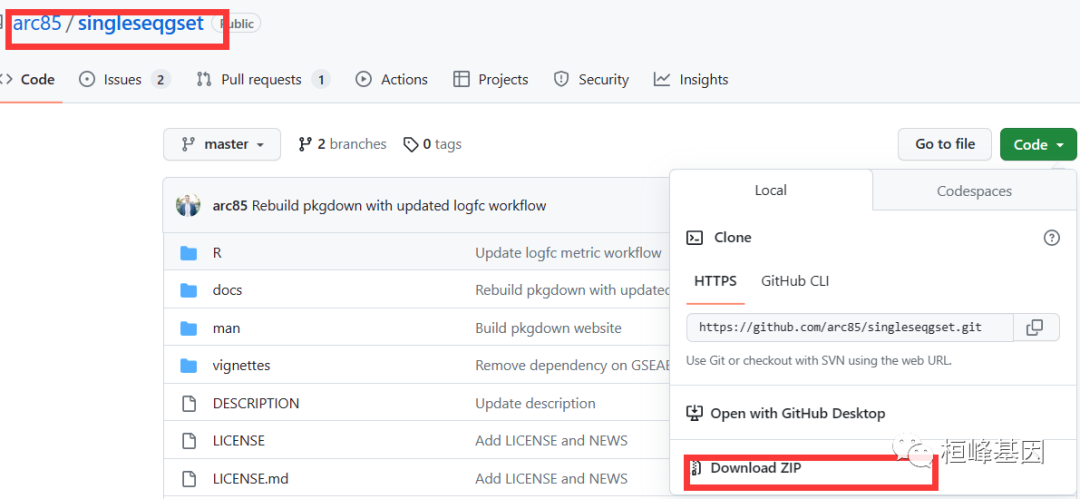
2. 在软件包里面直接复制出来如下两个R脚本。
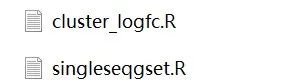
3. 使用 Rstudio 打开 "cluster_logfc.R"脚本文件,找到需要修改的字段,直接修改即可。
4. 使用 Rstudio 打开 "wmw_gsea.R"脚本文件,找到需要修改的字段,直接修改即可。
5. 保存之后,可以通过source()的方式直接调取脚本,如下:
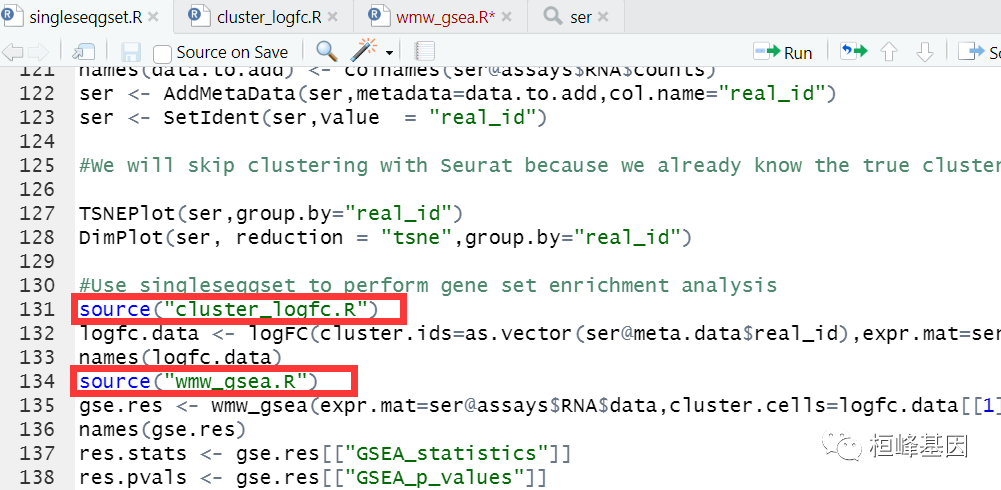
也可以再次打包为软件包,本地加载即可。
桓峰基因,铸造成功的您!
未来桓峰基因公众号将不间断的推出单细胞系列生信分析教程,
敬请期待!!
有想进生信交流群的老师可以扫最后一个二维码加微信,备注“单位+姓名+目的”!!!

桓峰基因官网正式上线,请大家多多关注,还有很多不足之处,大家多多指正!
http://www.kyohogene.com/
桓峰基因和投必得合作,文章润色优惠85折,需要文章润色的老师可以直接到网站输入领取桓峰基因专属优惠券码:KYOHOGENE,然后上传,付款时选择桓峰基因优惠券即可享受85折优惠哦!https://www.topeditsci.com/