
–https://stuartlab.org/signac/articles/pbmc_vignette
好的,开始学习scATAC-seq的数据是怎么玩的了,先跑完Signac的教程,边跑边思考怎么跟自己的课题相结合。
留意更多内容,欢迎关注微信公众号:组学之心
数据和R包的准备
数据下载:
wget https://cf.10xgenomics.com/samples/cell-atac/1.0.1/atac_v1_pbmc_10k/atac_v1_pbmc_10k_filtered_peak_bc_matrix.h5
wget https://cf.10xgenomics.com/samples/cell-atac/1.0.1/atac_v1_pbmc_10k/atac_v1_pbmc_10k_singlecell.csv
wget https://cf.10xgenomics.com/samples/cell-atac/1.0.1/atac_v1_pbmc_10k/atac_v1_pbmc_10k_fragments.tsv.gz
wget https://cf.10xgenomics.com/samples/cell-atac/1.0.1/atac_v1_pbmc_10k/atac_v1_pbmc_10k_fragments.tsv.gz.tbi
R包下载:
install.packages("Signac")if (!requireNamespace("EnsDb.Hsapiens.v75", quietly = TRUE))BiocManager::install("EnsDb.Hsapiens.v75")
此外还要准备好Seurat包
1.数据的预处理
Signac要用到两个文件:
-
Peak/Cell matrix:这类似于用于分析单细胞 RNA 测序的基因表达count矩阵。但是,矩阵的每一行代表的不是基因,而是基因组的一个区域(peak),预计代表开放染色质的一个区域。矩阵中的每个值代表每个单个条形码(即细胞)在每个峰内映射的 Tn5 整合位点的数量。
-
Fragment file:这代表所有单个细胞中所有唯一片段的完整列表。它是一个相当大的文件,处理速度较慢,并且存储在磁盘上(而不是内存中)。此文件它包含与每个单个细胞相关的所有片段,而不是仅包含映射到峰的片段。
Peak/Cell matrix读入
counts <- Read10X_h5(filename = "00practice/PBMC10k_scATAC/atac_v1_pbmc_10k_filtered_peak_bc_matrix.h5")
counts[1:4, 1:4]
dim(counts)
counts矩阵是稀疏矩阵,它大致长这样,行是染色质开放的区域,列是细胞ID:
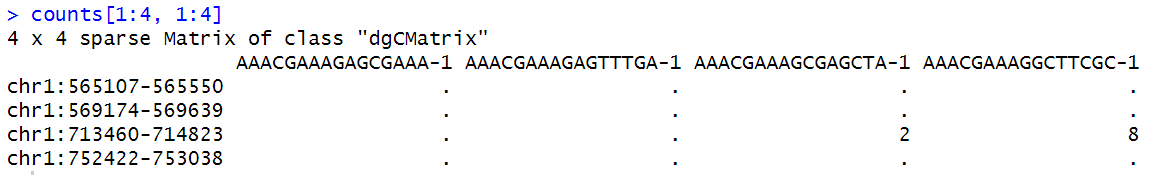
89796个开放区 8728个细胞
metadata读入
metadata <- read.csv(file = "00practice/PBMC10k_scATAC/atac_v1_pbmc_10k_singlecell.csv",header = TRUE, row.names = 1)
colnames(metadata)
metadata里有16个变量,它们的含义如下:
-
total: 总共测序到的片段数,包括所有读数,无论它们是否通过过滤。
-
duplicate: 标记为重复的片段数量。重复片段通常是由于 PCR 扩增引起的,它们不反映实际的生物学信号,因此通常会被过滤掉。
-
chimeric: 嵌合片段的数量。这些片段可能是在测序过程中由不同的 DNA 片段错误结合而成,通常会被过滤掉。
-
unmapped: 未能映射到参考基因组的片段数量。这些片段可能由于质量低或其他原因未能匹配到参考序列。
-
lowmapq: 映射质量(MAPQ)值低的片段数量。MAPQ 值低的片段通常指示不可靠的映射结果。
-
mitochondrial: 映射到线粒体基因组的片段数量。在线粒体富集的情况下,这一项可能较高。
-
passed_filters: 通过所有过滤标准的片段数量。这些片段通常被认为是高质量的片段,适合下游分析。
-
cell_id: 每个单细胞的唯一标识符,通常是细胞条形码。
-
is__cell_barcode: 指示某个条形码是否被标记为细胞条形码。0和1来表示,1表明该条形码是否通过了细胞条形码的过滤标准。
-
TSS_fragments: 转录起始位点(TSS)附近的片段数量。这些片段有助于识别基因表达的潜在调控区域。
-
DNase_sensitive_region_fragments: 映射到 DNase I 敏感区域的片段数量。这些区域通常是染色质开放的,可能包含调控元件。
-
enhancer_region_fragments: 增强子区域的片段数量。增强子是调控基因表达的重要元件。
-
promoter_region_fragments: 启动子区域的片段数量。启动子是转录起始的关键区域,常与基因表达的调控有关。
-
on_target_fragments: 在预期靶区域内的片段数量。靶区域通常是指在实验设计中特别关注的基因组区域。
-
blacklist_region_fragments: 映射到基因组黑名单区域的片段数量。这些区域通常是已知的异常区域(如高噪声区域)且通常在分析中被过滤掉。
-
peak_region_fragments: 映射到信号峰值区域的片段数量。信号峰值通常代表染色质开放区域,这些数据用于识别可能的调控元件。
Fragment文件读入并构建seurat项目
chrom_assay <- CreateChromatinAssay(counts = counts, sep = c(":", "-"),fragments = '00practice/PBMC10k_scATAC/atac_v1_pbmc_10k_fragments.tsv.gz',min.cells = 10, min.features = 200)pbmc <- CreateSeuratObject(counts = chrom_assay, assay = "peaks", meta.data = metadata)
pbmc## An object of class Seurat
## 87561 features across 8728 samples within 1 assay
## Active assay: peaks (87561 features, 0 variable features)
## 2 layers present: counts, data
文件内容长得很像scRNA-seq,但metadata有很大不一样
ATAC-seq 数据使用custom assay ChromatinAssay 进行存储。这启用了一些专门用于分析基因组单细胞检测(例如 scATAC-seq)的功能。通过打印assay,我们可以看到 ChromatinAssay 中可以包含的一些其他信息,包括基序信息、基因注释和基因组信息。
## ChromatinAssay data with 87561 features for 8728 cells
## Variable features: 0
## Genome:
## Annotation present: FALSE
## Motifs present: FALSE
## Fragment files: 1
添加基因注释
# 下载获取基因注释文件,包含有关基因组位置的信息,
#例如染色体、起始和结束位置,以及相关的元数据
annotations <- GetGRangesFromEnsDb(ensdb = EnsDb.Hsapiens.v75)
# 给染色体名称加个chr前缀
seqlevels(annotations) <- paste0('chr', seqlevels(annotations))# 把annotations的基因组版本被设置为 "hg19"
genome(annotations) <- "hg19"Annotation(pbmc) <- annotations
pbmc的assay-peaks中多了个annotations

2.计算质控指标
Signac作者建议使用以下指标来质控:
-
①Nucleosome banding pattern:DNA 片段大小的直方图(由双端测序读数确定)应显示与缠绕单个核小体的 DNA 长度相对应的强核小体带型。在每个单细胞计算它,并量化单核小体片段与无核小体片段的近似比率(存储为 nucleosome_signal)
-
②Transcriptional start site (TSS) enrichment score:ENCODE 项目已根据以 TSS 为中心的片段与 TSS 侧翼区域中的片段的比率定义了 ATAC-seq 靶向分数。较差的 ATAC-seq 实验通常会具有较低的 TSS 富集分数。我们可以使用 TSSEnrichment() 函数为每个细胞计算此指标,结果存储在列名 TSS.enrichment 下的元数据中。
-
③Total number of fragments in peaks:细胞测序深度/复杂性的量度。由于测序深度低,可能需要排除读取量非常少的细胞。水平极高的细胞可能代表双细胞、细胞核团块或其他伪影。
-
④Fraction of fragments in peaks:表示落在 ATAC-seq 峰内的所有片段的分数。值较低的细胞(即 <15-20%)通常代表应移除的低质量细胞或技术伪影。请注意,此值可能对使用的峰集很敏感。
-
⑤Ratio reads in genomic blacklist regions:ENCODE 项目提供了黑名单区域列表,表示通常与伪影信号相关的读取。与映射到峰的读取相比,映射到这些区域的读取比例较高的细胞通常代表技术伪影,应将其移除。 Signac 包中包含人类 (hg19 和 GRCh38)、小鼠 (mm10)、果蝇 (dm3) 和秀丽隐杆线虫 (ce10) 的 ENCODE 黑名单区域。
# 计算每个细胞的nucleosome signal值
pbmc <- NucleosomeSignal(object = pbmc)# 计算每个细胞的TSS enrichment值
pbmc <- TSSEnrichment(object = pbmc, fast = FALSE)# 添加黑名单比例和峰值读取分数
pbmc$pct_reads_in_peaks <- pbmc$peak_region_fragments / pbmc$passed_filters * 100
pbmc$blacklist_ratio <- pbmc$blacklist_region_fragments / pbmc$peak_region_fragments

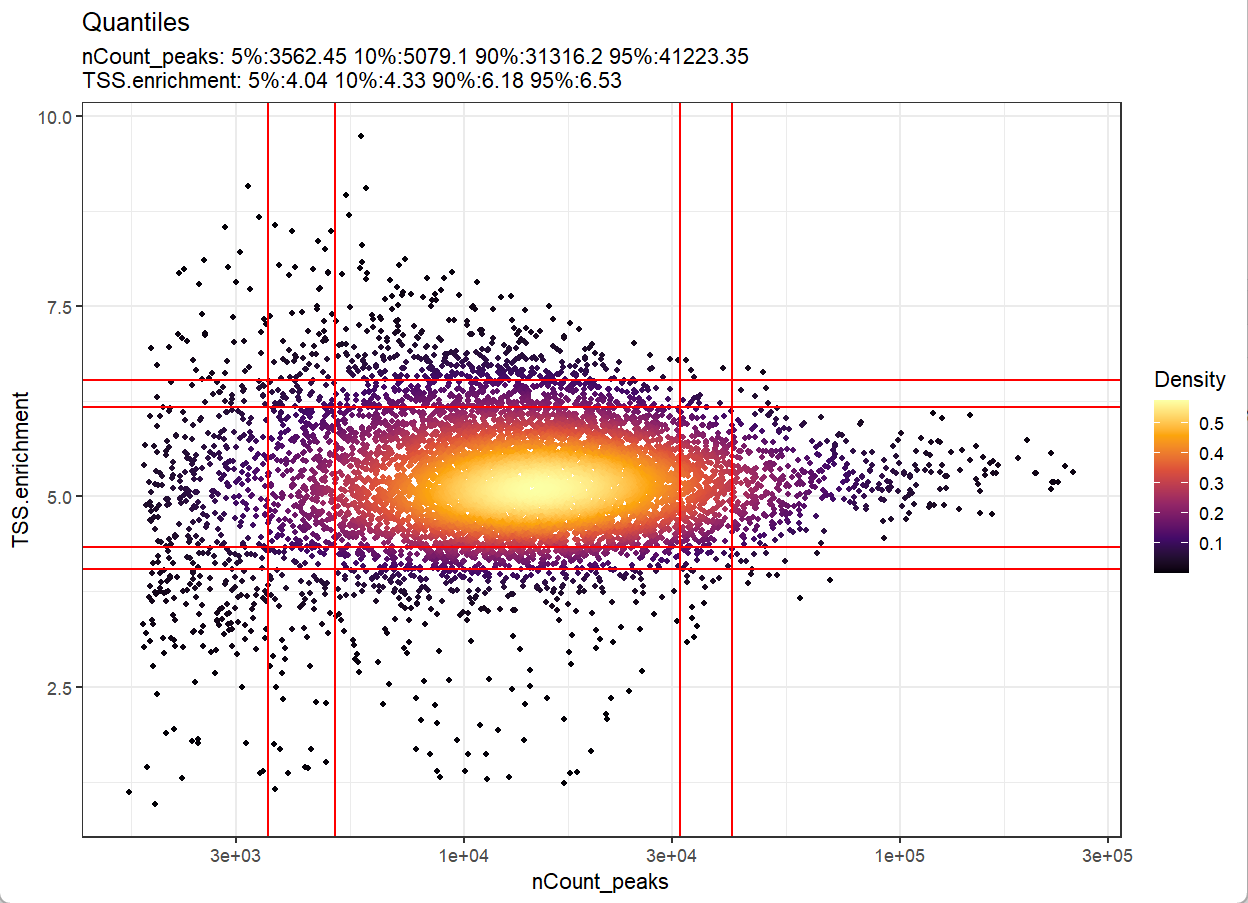
可以使用 DensityScatter() 函数可视化存储在项目中metadata中的变量之间的关系。通过设置 quantiles=TRUE,还可以快速找到不同 QC 指标的合适截止值:
DensityScatter(pbmc, x = 'nCount_peaks', y = 'TSS.enrichment', log_x = TRUE, quantiles = TRUE)
使用 TSSPlot() 函数对不同细胞组的 TSS 富集信号进行下游绘制
pbmc$high.tss <- ifelse(pbmc$TSS.enrichment > 3, 'High', 'Low')
TSSPlot(pbmc, group.by = 'high.tss') + NoLegend()

上图反映了在转录起始位点附近的染色质开放程度。高 TSS 富集分数通常表示在这些区域有更多的开放染色质,这对于识别调控元件和基因表达调控非常重要。通过 pbmc$TSS.enrichment > 3 这个条件,数据被分为 “High” 和 “Low” 两个组。这个阈值用于区分在 TSS 区域附近有高水平和低水平开放染色质的细胞群体。
pbmc$nucleosome_group <- ifelse(pbmc$nucleosome_signal > 4, 'NS > 4', 'NS < 4')
FragmentHistogram(object = pbmc, group.by = 'nucleosome_group')

左图 (“NS < 4”):显示了核小体信号较低的细胞群体中片段长度的分布。可以看到,这些细胞中短片段(通常 < 100 bp)占比较高,这些短片段多为无核小体片段,通常反映了开放的染色质区域。
右图 (“NS > 4”):显示了核小体信号较高的细胞群体中片段长度的分布。在这部分细胞中,片段长度的分布有一个在 150-200 bp 范围内的明显峰值,这是由于核小体包裹的 DNA 片段长度大约在这个范围。
核小体信号较高的群体通常表明这些细胞中较多的 DNA 被核小体包裹,而核小体信号较低的群体则表明染色质可能较为开放。
VlnPlot(object = pbmc, features = c('nCount_peaks', 'TSS.enrichment', 'blacklist_ratio', 'nucleosome_signal', 'pct_reads_in_peaks'),pt.size = 0.1,ncol = 5)
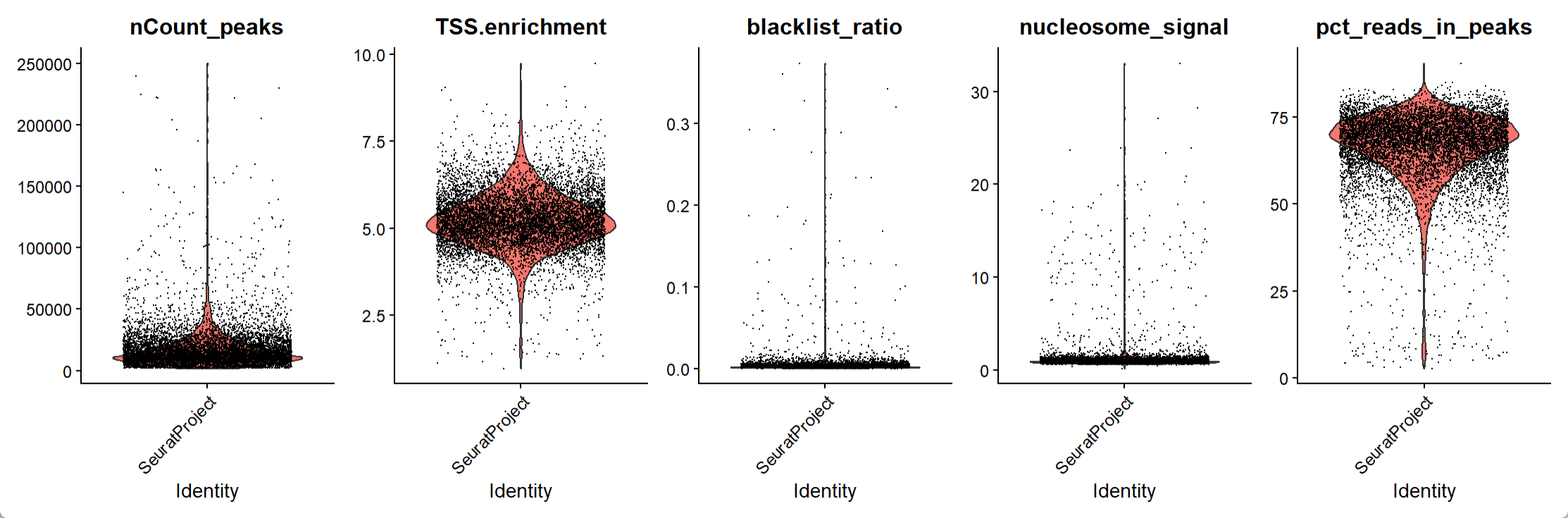
小提琴图展示出刚刚说的5个指标的统计分布。用以下阈值来筛选(没有最优解,只有适合与否):
pbmc <- subset(x = pbmc,subset = nCount_peaks > 3000 &nCount_peaks < 30000 &pct_reads_in_peaks > 15 &blacklist_ratio < 0.05 &nucleosome_signal < 4 &TSS.enrichment > 3
)
pbmc## An object of class Seurat
## 87561 features across 7307 samples within 1 assay
## Active assay: peaks (87561 features, 0 variable features)
## 2 layers present: counts, data
3.归一化和线性降维
-
归一化:Signac 执行term frequency-inverse document frequency (TF-IDF) 归一化。这是一个两步归一化过程,既跨细胞归一化以校正细胞测序深度的差异,又跨峰值归一化以赋予更罕见的峰值更高的值。
-
特征选择:scATAC-seq 数据的低动态范围使得执行变量特征选择变得具有挑战性。相反,我们可以选择仅使用前 n% 的特征(峰值)进行降维,或者使用 FindTopFeatures() 函数删除少于 n 个细胞中存在的特征。在这里将使用所有特征。此函数会自动将用于降维的特征设置为 Seurat 项目的 VariableFeatures()。
-
降维:使用上面选择的特征(峰值)对 TD-IDF 矩阵进行奇异值分解 (SVD),将返回对象的降维表示。
pbmc <- RunTFIDF(pbmc)
pbmc <- FindTopFeatures(pbmc, min.cutoff = 'q0')
pbmc <- RunSVD(pbmc)
TF-IDF 和 SVD 的组合步骤称为潜在语义索引 (LSI)。第一个 LSI component 通常捕获测序深度(技术变化)而不是生物变化。如果是这种情况,则应从下游分析中删除该组件。我们可以使用 DepthCor() 函数评估每个 LSI 组件与测序深度之间的相关性:
DepthCor(pbmc)
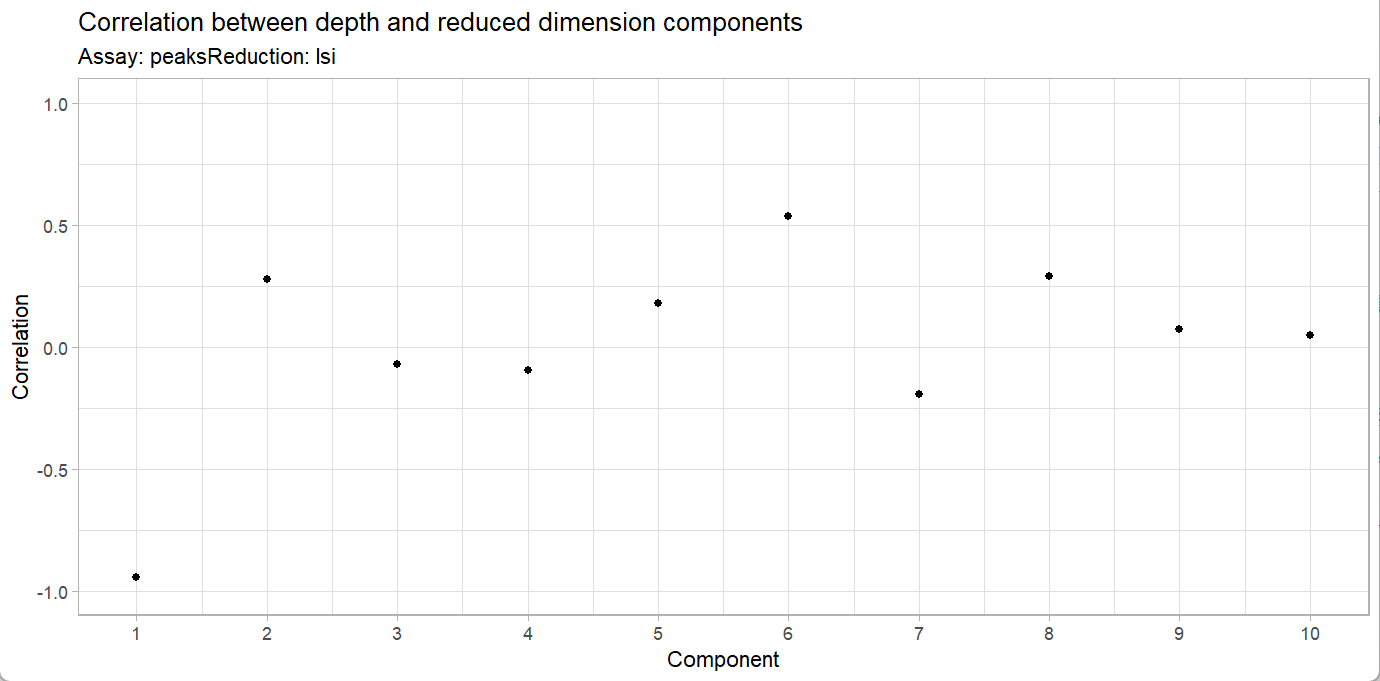
可以看到第一个 LSI 成分和细胞的总计数数之间存在非常强的相关性,把它不纳入下游分析中。
4.非线性降维和聚类
pbmc <- RunUMAP(object = pbmc, reduction = 'lsi', dims = 2:30)
pbmc <- FindNeighbors(object = pbmc, reduction = 'lsi', dims = 2:30)
pbmc <- FindClusters(object = pbmc, verbose = FALSE, algorithm = 3)
DimPlot(object = pbmc, label = TRUE) + NoLegend()
注意这里的dims选择是第二个开始。

5.创建基因活性矩阵
在 scATAC-seq 数据中注释和解释簇更具挑战性,因为我们对非编码基因组区域的功能作用的了解远少于对蛋白质编码区域(基因)的了解。
我们可以通过评估与基因相关的染色质可及性来量化基因组中每个基因的活性,并创建一个源自 scATAC-seq 数据的新基因活性检测。在这里使用一种简单的方法来求和基因体和启动子区域相交的片段。
为了创建基因活动矩阵,先提取基因坐标并将其扩展为包含 2 kb 上游区域(因为启动子的可及性通常与基因表达相关)。然后,我们使用 FeatureMatrix() 函数计算映射到每个区域的每个细胞的片段数。这些步骤由 GeneActivity() 函数自动执行:
gene.activities <- GeneActivity(pbmc)# 将基因活动矩阵作为新检测添加到 Seurat 对象并对其进行归一化
pbmc[['RNA']] <- CreateAssayObject(counts = gene.activities)
pbmc <- NormalizeData(object = pbmc,assay = 'RNA',normalization.method = 'LogNormalize',scale.factor = median(pbmc$nCount_RNA)
)
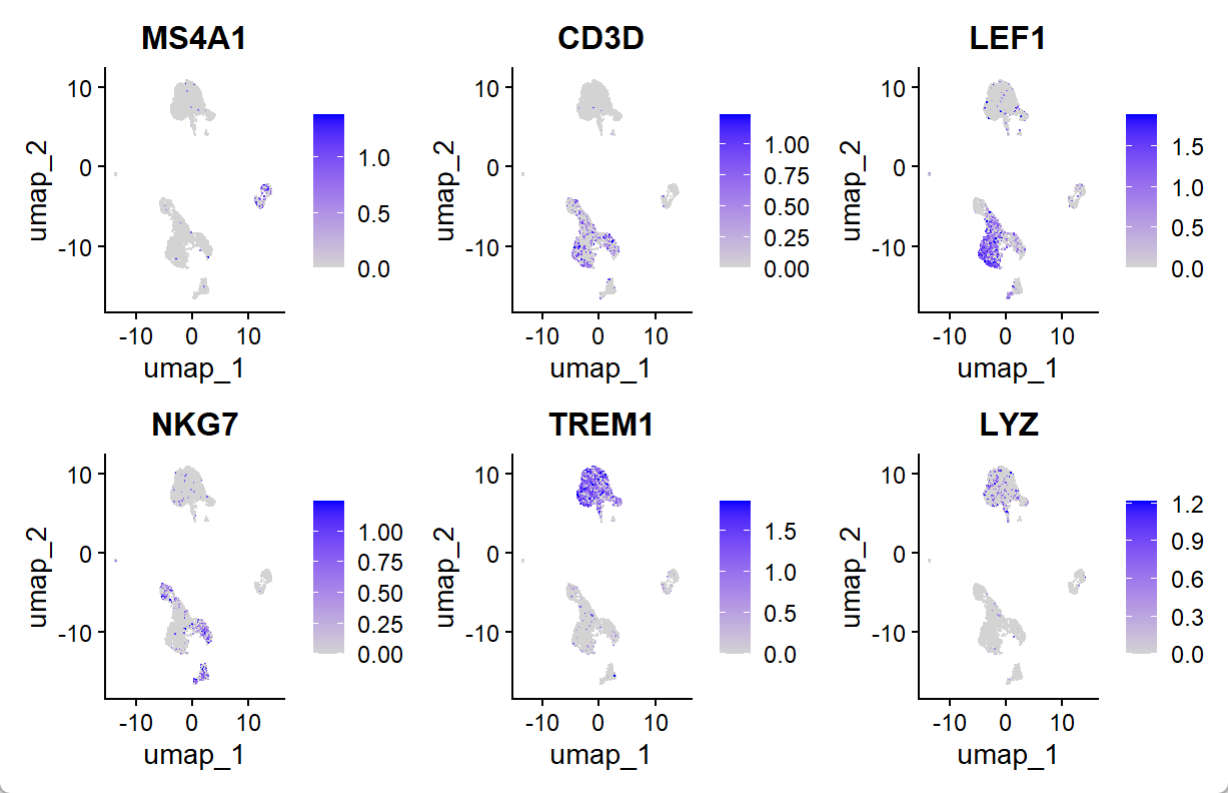
现在可以将典型标记基因的活性可视化,以帮助解释我们的 ATAC-seq 聚类结果。这些活性将比 scRNA-seq 测量值更嘈杂,因为它们代表了稀疏染色质数据的测量值,并且它们假设基因体/启动子的可及性和基因表达之间存在一般对应关系,但情况可能并非总是如此。但还是可以根据这些基因活动概况开始辨别单核细胞、B、T 和 NK 细胞的群体。
DefaultAssay(pbmc) <- 'RNA'FeaturePlot(object = pbmc,features = c('MS4A1', 'CD3D', 'LEF1', 'NKG7', 'TREM1', 'LYZ'),pt.size = 0.1,max.cutoff = 'q95',ncol = 3
)
6.整合scRNA-seq数据
为了帮助解释 scATAC-seq 数据,可以根据来自同一类型样本的 scRNA-seq 对细胞进行分类。并跨模态学习基因活性矩阵和 scRNA-seq 数据集中的共享相关模式,以识别两种模态之间匹配的生物状态。此过程为每个 scRNA-seq 定义的簇标签返回每个细胞的分类值。
读取pbmc的rna测序数据
pbmc_rna <- readRDS("00practice/PBMC10k_scATAC/pbmc_10k_v3.rds")
pbmc_rna <- UpdateSeuratObject(pbmc_rna)
找到转移锚点transfer anchors
transfer.anchors <- FindTransferAnchors(reference = pbmc_rna,query = pbmc,reduction = 'cca'
)predicted.labels <- TransferData(anchorset = transfer.anchors,refdata = pbmc_rna$celltype,weight.reduction = pbmc[['lsi']],dims = 2:30
)pbmc <- AddMetaData(object = pbmc, metadata = predicted.labels)
分类值:
FindTransferAnchors 函数被用来在两个不同的单细胞数据集之间找到转移锚点(transfer anchors)。这些锚点帮助将信息从一个参考数据集(如单细胞 RNA 测序数据)转移到另一个查询数据集(如 scATAC-seq 数据),以推断在没有 RNA 数据的情况下细胞的基因表达。这对于跨数据类型的单细胞分析非常有用,有助于整合和解释多种数据源的信息。
TransferData 函数将细胞类型注释或其他信息从参考数据集传递到查询数据集中。
作图展示效果
plot1 <- DimPlot(object = pbmc_rna,group.by = 'celltype',label = TRUE,repel = TRUE) + NoLegend() + ggtitle('scRNA-seq')plot2 <- DimPlot(object = pbmc,group.by = 'predicted.id',label = TRUE,repel = TRUE) + NoLegend() + ggtitle('scATAC-seq')plot1 + plot2

可见基于 scRNA 的分类与使用 scATAC-seq 数据计算的 UMAP 可视化一致。为了结合我们的 scATAC-seq 聚类和标签转移结果,我们可以将簇名称重新分配给该簇最常见的预测标签。
for(i in levels(pbmc)) {cells_to_reid <- WhichCells(pbmc, idents = i)newid <- names(which.max(table(pbmc$predicted.id[cells_to_reid])))Idents(pbmc, cells = cells_to_reid) <- newid
}
7.查找不同细胞类型之间差异可及的peak
计算差异可及性peak
为了找到细胞簇之间的差异可及区域,需要执行差异可及性 (DA) 测试。方法1:一种简单的方法是执行 Wilcoxon 秩和检验,presto 包已经实现了可以在 Seurat 对象上运行的极快 Wilcoxon 检验。
方法2:利用逻辑回归,将片段总数作为潜在变量添加,以减轻差异测序深度对结果的影响。在这里重点比较 Naive CD4 细胞和 CD14 单核细胞,但可以使用这些方法比较任何细胞组。我们还可以在 Seurat 中的小提琴图、特征图、点图、热图或任何可视化工具上可视化这些marker peaks。
这里使用方法2来执行:
DefaultAssay(pbmc) <- 'peaks'da_peaks <- FindMarkers(object = pbmc,ident.1 = "CD4 Naive",ident.2 = "CD14+ Monocytes",test.use = 'LR',latent.vars = 'nCount_peaks'
)
head(da_peaks)## p_val avg_log2FC pct.1 pct.2 p_val_adj
## chr14-99721608-99741934 1.414340e-280 5.571161 0.868 0.022 1.238410e-275
## chr14-99695477-99720910 5.529507e-222 5.100310 0.797 0.021 4.841691e-217
## chr17-80084198-80086094 8.962660e-221 7.032939 0.668 0.005 7.847795e-216
## chr7-142501666-142511108 6.506936e-212 4.757277 0.754 0.029 5.697538e-207
## chr2-113581628-113594911 5.746054e-188 -5.051770 0.035 0.663 5.031302e-183
## chr6-44025105-44028184 2.328105e-179 -4.394199 0.046 0.616 2.038512e-174
plot1 <- VlnPlot(object = pbmc,features = rownames(da_peaks)[1],pt.size = 0.1,idents = c("CD4 Naive","CD14+ Monocytes")
)
plot2 <- FeaturePlot(object = pbmc,features = rownames(da_peaks)[1],pt.size = 0.1
)plot1 | plot2
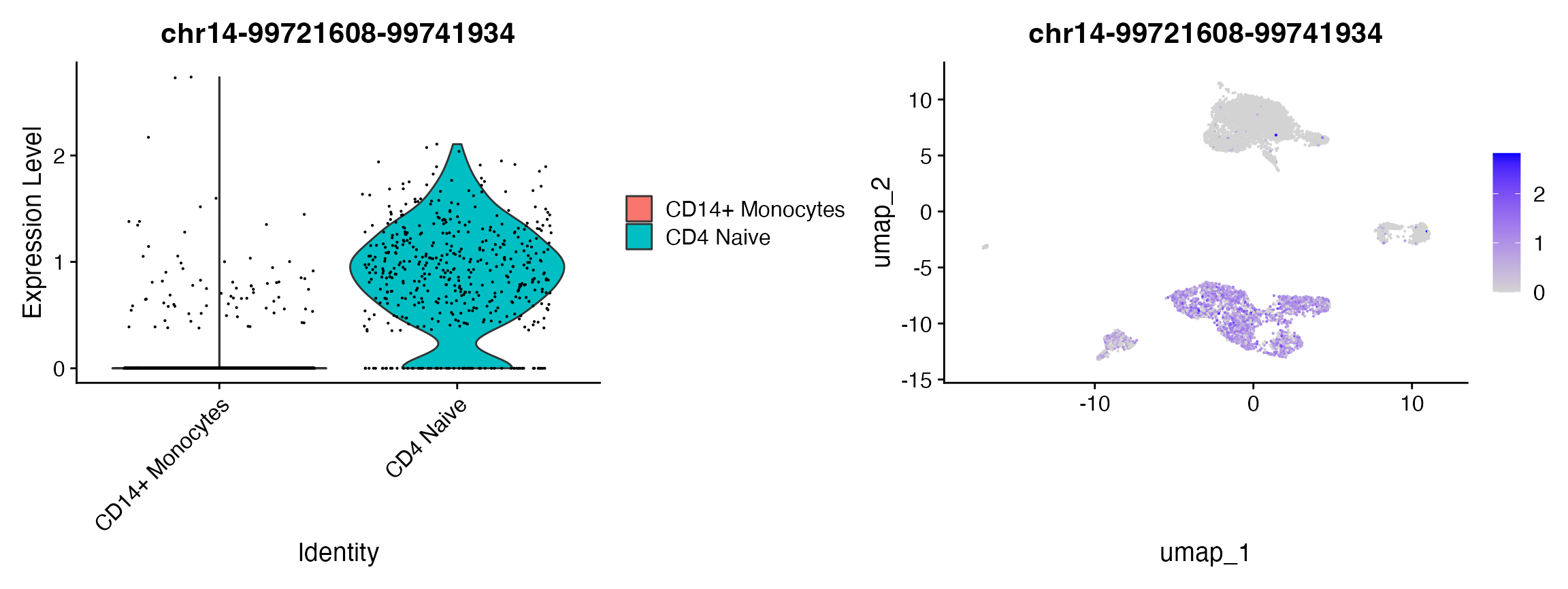
另一种查找两组细胞之间 DA 区域的方法是查看两组细胞之间的倍数变化可及性。这比运行更复杂的 DA 测试要快得多,但无法解释潜在变量,例如细胞之间总测序深度的差异,并且不执行任何统计测试。然而,这仍然是一种快速探索数据的有效方法,可以使用 Seurat 中的 FoldChange() 函数来执行。
fc <- FoldChange(pbmc, ident.1 = "CD4 Naive", ident.2 = "CD14+ Monocytes")
# order by fold change
fc <- fc[order(fc$avg_log2FC, decreasing = TRUE), ]
head(fc)## avg_log2FC pct.1 pct.2
## chr6-28416849-28417227 11.45411 0.067 0
## chr7-110665002-110665493 11.41080 0.073 0
## chr8-19317420-19317942 11.22146 0.061 0
## chr1-172836553-172836955 11.20312 0.058 0
## chr2-191380525-191380926 11.15811 0.056 0
## chr8-90255778-90256179 11.03921 0.050 0
找到距离每个峰值最近的基因
峰值可能难以单独解释。我们可以使用 ClosestFeature() 函数找到距离每个峰值最近的基因。如果你探索基因列表,你会发现幼稚 T 细胞中打开的峰值接近 BCL11B 和 GATA3(T 细胞分化的关键调节因子)等基因,而单核细胞中打开的峰值接近 CEBPB(单核细胞分化的关键调节因子)等基因。我们可以通过对 ClosestFeature() 返回的基因集进行基因本体富集分析来进一步跟踪这一结果,有许多 R 包可以做到这一点(例如,GOstats 包)。
open_cd4naive <- rownames(da_peaks[da_peaks$avg_log2FC > 3, ])
open_cd14mono <- rownames(da_peaks[da_peaks$avg_log2FC < -3, ])closest_genes_cd4naive <- ClosestFeature(pbmc, regions = open_cd4naive)
closest_genes_cd14mono <- ClosestFeature(pbmc, regions = open_cd14mono)head(closest_genes_cd4naive)## tx_id gene_name gene_id gene_biotype type
## ENST00000443726 ENST00000443726 BCL11B ENSG00000127152 protein_coding cds
## ENST00000357195 ENST00000357195 BCL11B ENSG00000127152 protein_coding cds
## ENST00000583593 ENST00000583593 CCDC57 ENSG00000176155 protein_coding cds
## ENSE00002456092 ENST00000463701 PRSS1 ENSG00000204983 protein_coding exon
## ENST00000546420 ENST00000546420 CCDC64 ENSG00000135127 protein_coding cds
## ENST00000455990 ENST00000455990 HOOK1 ENSG00000134709 protein_coding cds
## closest_region query_region distance
## ENST00000443726 chr14-99737498-99737555 chr14-99721608-99741934 0
## ENST00000357195 chr14-99697682-99697894 chr14-99695477-99720910 0
## ENST00000583593 chr17-80085568-80085694 chr17-80084198-80086094 0
## ENSE00002456092 chr7-142460719-142460923 chr7-142501666-142511108 40742
## ENST00000546420 chr12-120427684-120428101 chr12-120426014-120428613 0
## ENST00000455990 chr1-60280790-60280852 chr1-60279767-60281364 0
head(closest_genes_cd14mono)## tx_id gene_name gene_id gene_biotype
## ENST00000432018 ENST00000432018 IL1B ENSG00000125538 protein_coding
## ENSE00001638912 ENST00000455005 RP5-1120P11.3 ENSG00000231881 lincRNA
## ENST00000445003 ENST00000445003 RP11-290F20.3 ENSG00000224397 lincRNA
## ENST00000568649 ENST00000568649 PPCDC ENSG00000138621 protein_coding
## ENST00000409245 ENST00000409245 TTC7A ENSG00000068724 protein_coding
## ENST00000484822 ENST00000484822 RXRA ENSG00000186350 protein_coding
## type closest_region query_region distance
## ENST00000432018 cds chr2-113593760-113593806 chr2-113581628-113594911 0
## ENSE00001638912 exon chr6-44041650-44042535 chr6-44025105-44028184 13465
## ENST00000445003 gap chr20-48884201-48894027 chr20-48889794-48893313 0
## ENST00000568649 cds chr15-75335782-75335877 chr15-75334903-75336779 0
## ENST00000409245 cds chr2-47300841-47301062 chr2-47297968-47301173 0
## ENST00000484822 gap chr9-137211331-137293477 chr9-137263243-137268534 0
8.绘制基因组区域
我们可以使用 CoveragePlot() 函数绘制按簇、细胞类型或对象中存储的任何其他metadata分组的细胞基因组区域 Tn5 酶整合频率。这些代表pseudo-bulk可访问性轨迹,其中组内所有细胞的信号被平均在一起以可视化区域内的 DNA 可访问性。除了这些可访问性轨迹外,我们还可以可视化其他重要信息,包括基因注释、峰值坐标和基因组链接。
levels(pbmc) <- c("CD4 Naive","CD4 Memory","CD8 Naive","CD8 effector","Double negative T cell","NK dim", "NK bright", "pre-B cell",'B cell progenitor',"pDC","CD14+ Monocytes",'CD16+ Monocytes')CoveragePlot(object = pbmc,region = rownames(da_peaks)[1],extend.upstream = 40000,extend.downstream = 20000
)
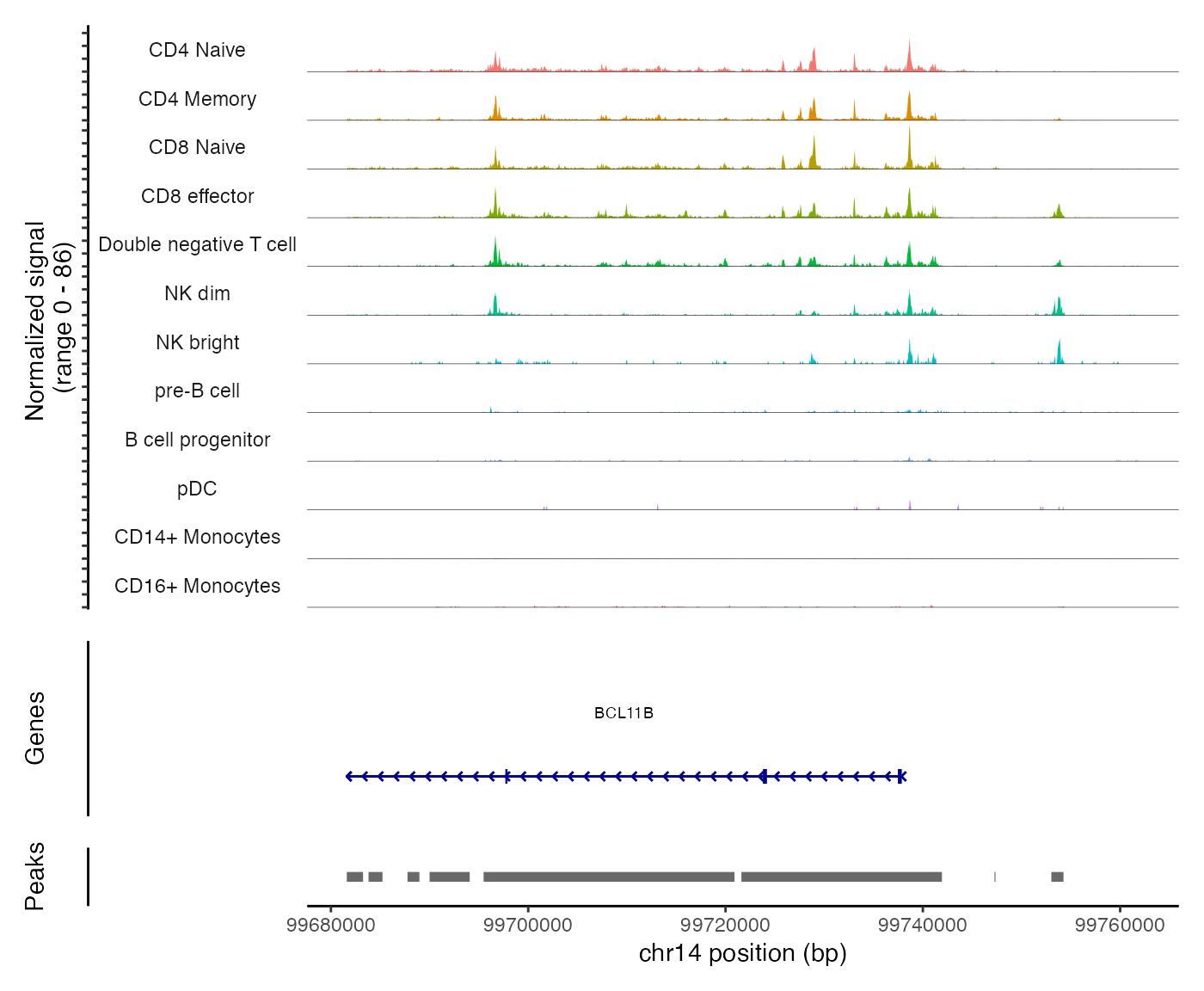
我们还可以使用 CoverageBrowser() 函数创建这些图的交互式版本:
p.list <- CoverageBrowser(object = pbmc, region = "CD8A")

可以自己任意修改这里的选项来展示基因组区域。
基本的流程是这样,上手试试吧!




