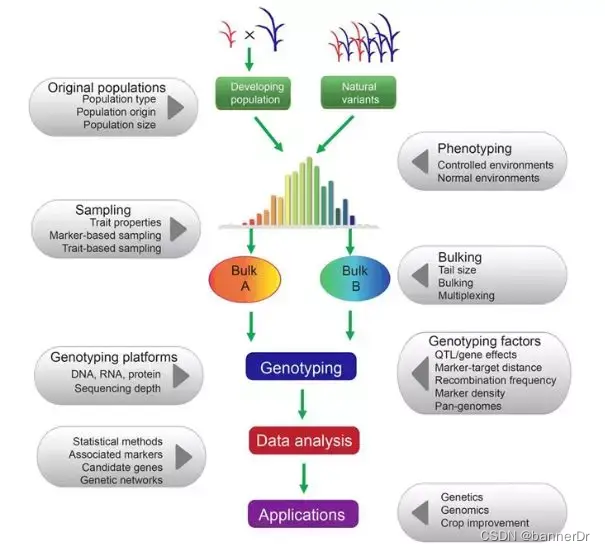
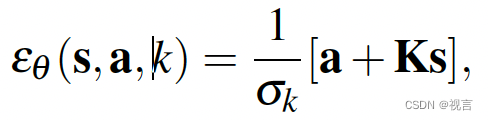1、混合(Pooling)样本测序研究
https://www.jianshu.com/p/19ce438ccccf
1.1 混合测序基础
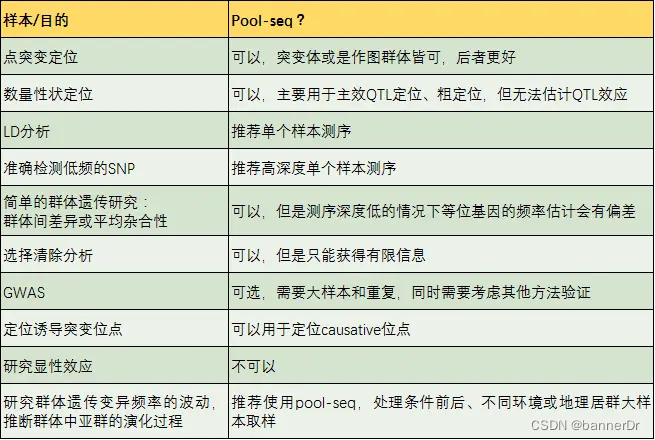
测序成本虽然下降了,但对于植物育种应用研究来说还是很高,动不动就上百群体,小小植物个体价值又低,测完了很可能后面就用不到了。这时,混合样本测序是一种省钱的好办法。
混池测序(Pool-seq)相对于GWAS或其他精细定位策略而言,其实是一个初定位产品,其结果很有可能是跟性状相关的候选区域。
概念:
混合样本测序一般是选择表型极端或目标性状差异的个体混合,构建一个文库进行测序。
原理:
假设每个样本被测到的概率相等,通过测序reads数计算等位基因频率。如果基因与研究性状有关,那么理想情况下,表型差异显著的混合样本中,该基因等位基因频率差异显著。
不足:
- 大群体的等位基因频率才能代表该群体真实的情况,选择少量样本可能带来选样误差;
- 各样本测序量不均一引入新的偏差。
但研究表明,在大样本量混合且提高测序深度的情况下,混合样本能够准确评估等位基因频率。
影响因素及建议:
-
群体类型:群体类型决定研究背景是否纯,影响定位的精确性。混合样本测序最好是只有目标性状存在差异,其他性状一致,即遗传背景纯,一般永久群体>临时群体>自然群体。
-
混合样本量:多态性高的群体(如F2),推荐混合样本量>100;多态性低的群体(如BCF),推荐混合样本量>20;且作图群体选择比例<25%。
-
亲本选择:两个亲本尽量性状差异单一,杂合位点少。
-
混合样本的均一性:样本量小的时候影响大,样本量大影响小。
-
表型:表型统计不准确,或由多个微效基因控制,会引起定位效果不佳。
-
参考基因组:基因组组装好坏,基因组注释情况,物种连锁不平衡强易导致候选区域过大。建议采用组装到染色体水平的参考基因组。
-
测序错误:混合样本测序比较难通过算法区分是测序错误还是稀有变异,测序深度高能有效降低影响。
-
测序数据量:测序数据量推荐50X以上,测序深度高有利于检测到多态的SNP位点。
-
比对:混合样本无法校正比对错误,CNV会影响等位基因频率统计。
1.2 点突变检测
对于隐形纯合点突变,效果较好。
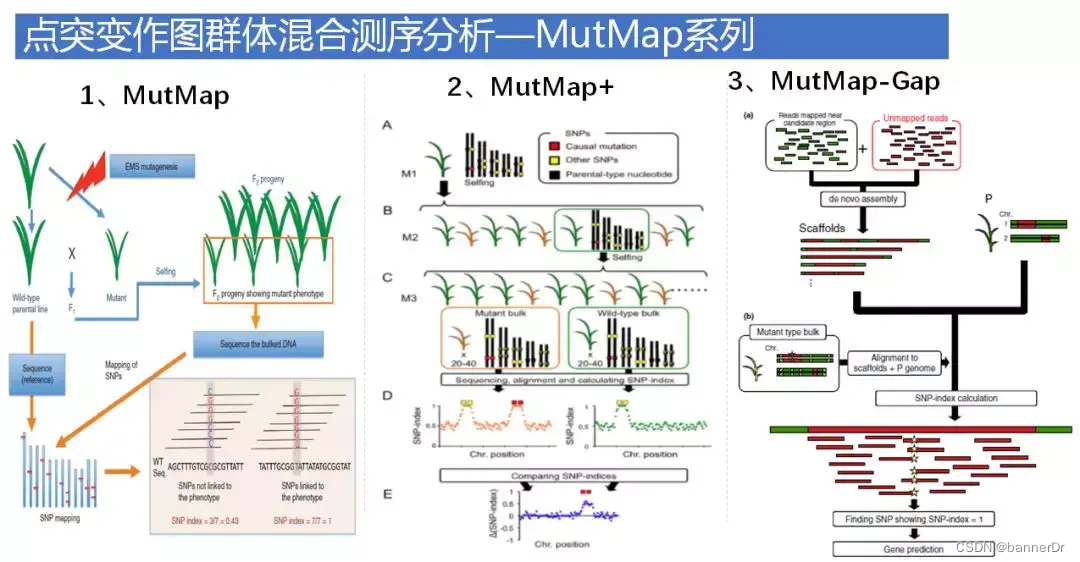
MutMap和MutMap+是利用SNP-index算法,需要参考基因组,如果目标位点位于参考基因组没有组装上的gap区,或是参考基因组不具有的序列中,利用MutMap检测方法就不能有效检测到目标突变位点。
MutMap-Gap方法结合了MutMap和de novo组装。先通过MutMap分析SNP-index peak区,发现找不到跟突变性状相关的基因,再将之前比对不上参考基因组的野生型亲本unmapped reads和MutMap分析中SNP-index peak区域的野生型亲本比对上的reads一起进行de novo组装,获得潜在的新基因,并以此为参考再计算SNP-index,检测目标突变位点。
1.3 BSA
BSA(Bulked segregant analysis,混合分组分析),利用目标性状存在极端表型差异的两个亲本构建分离群体,在子代分离群体中,选取两组表型差异极端的个体分别构建混合池 ,结合高通量测序技术对混合样本测序,比较两组群体在多态位点(SNP)的等位基因频率(AF)是否具有显著差异,定位与目标性状相关联的位点并对其进行注释,研究控制目标性状的基因及其分子机制。
SNP-index是主流的BSA定位算法。其原理是构建子代分离群体,经过挑选极端性状构建混池后对SNP进行检测,对各混池进行等位基因频率分析,并与其中一个亲本进行比较。与此亲本不同的基因型所占的比例,即为该位点的SNP-index。
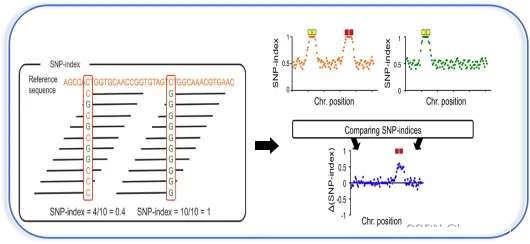
注意这里的reference并不是变异检测的参考基因组,而是构建群体所使用的亲本,所以SNP-index计算高度依赖于亲本测序数据。)
两个混池相减(上图右)得到了△SNP-index的结果,即两个混池之间SNP基因型频率的差异。理论上说,不与性状相关的位点,△SNP-index的值应当在0左右,代表混池之间不存在差异;而QTL及其相连锁位置的SNP,△SNP-index值应当呈现较高的数值。
△SNP index会存在因统计偏差造成的假阳性位点,可以通过计算滑窗内所有SNP的△SNP-index,来消除其影响,得到真正QTL所在的基因组区域。
其他算法如欧几里得距离(ED),Gradedpool-seq(Ridit检验)等。
这里的BSA是指狭义上的QTL-seq,针对有主效基因的数量性状。实际上上面的质量性状/点突变性状、InDel-seq(InDel突变性状)以及下面的BSR,都属于BSA的范畴,原理相似。此外还有QTG-seq。相应的Pipeline可参考:http://genome-e.ibrc.or.jp/home/bioinformatics-team/mutmap
1.4 BSR
BSR(Bulked segregant RNA sequencing)同样依据分组混合的原理,在RNA水平上进行高通量测序并定位候选基因,即BSA+RNAseq。BSR的混池同样选取分离群体中的极端性状单株,混池用的单株数会比BSA多一些(大多大于30),提取RNA进行混池,再进行转录组测序,mapping参考基因组后同样进行变异分析,确定候选区间。BSR的优势在于不仅提供变异信息,还能提供候选区域中基因的表达信息。
BSR的劣势:RNAseq只能检测表达基因上的SNP,检测的SNP数量少,一般只适用于高频的SNP。同时由于存在RNA编辑等问题,RNA层面检测的SNP和DNA层面也是有差别的,所以只有当DNA层面无法实现(复杂基因组)或DNA测序成本过高(超大基因组)等情况下可选择BSR,否则还是优先选择BSA。
1.5 混合样本GWAS分析
Pool –GWAS也是一种省钱策略,但还是非常小众。
比如:GWAS study using DNA pooling strategy identifies association of variant rs4910623 in OR52B4 gene with anti-VEGF treatment response in age-related macular degeneration
Pool –GWAS研究复杂遗传背景的性状功效降低,对稀有变异的检测能力下降。
1.6. 混合样本驯化研究
同样,分析获得的驯化相关位点很多,如果想用类似的方法检测复杂性状相关位点,后续挖掘真正的功能位点的难度还是很大。
1.7. 小结
2. BSA专题——分析方法大汇总 【派森诺】
https://www.sohu.com/a/414749205_120380672
在农业科学中,为了提升作物农艺性状,经常会遇到将与性状相关的基因或位点在基因组上进行定位的需求,此时BSA作为一种简便又高效的分析方法便有了大显身手的机会。可是BSA究竟是怎样的一种研究方法呢,适用于什么群体呢?跟着小编了解一下吧!
2.1 什么是BSA?
BSA(Bulked segregation analysis)即混合分组分析,也称分离群体分组分析,是指利用目标性状存在极端表型差异的两个亲本构建分离群体,在子代分离群体中,选取两组表型差异极端的个体分别构建混合池 ,结合高通量测序技术对混合样本测序,比较两组群体在多态位点(SNP)的等位基因频率(AF)是否具有显著差异,定位与目标性状相关联的位点并对其进行注释,研究控制目标性状的基因及其分子机制。
相较于传统的遗传学研究方法(基因定位常用分析方法,小编已经安排上啦!),BSA最大的特点是不需要对群体中的所有个体进行基因分型,而是对挑选的个体按照性状进行混合分析,所以可以极大地降低研究的工作量和成本。
2.2 什么样本适合BSA分析?
既然BSA已经兼具了简便,准确、高性价比等优点,自然也有自己的小性子了,BSA分析对使用的群体有一定要求。
1、 人工构建的遗传群体(最常用来的是F2、BC、RIL)。通常来说,使用自然群体和遗传群体都可以进行BSA分析,但是考虑到遗传背景较复杂,可能导致定位结果不理想,所以不推荐使用自然群体进行BSA研究。
2、 亲本目标性状差异明显,其他性状差异随机分布,所构建分离群体两个混池之间目标性状有显著差异,非目标性状无明显差异。
3、 有合适的参考基因组信息。参考基因组组装的越好,信息越全,对于后续基因定位和候选区间的注释都会更加精确,可以锁定候选区间并估计候选区域的大小。没有组装到染色体级别的参考基因组,分析思路是一样的,但只能得到某个或某些scaffolds中的snp与性状相关,无法估计候选区间大小,甚至再组装结果差的情况下,无法判断基因的物理位置。
2.3 BSA有哪些分析方法?
1、SNP index及△SNP index
SNP-index作为主流的BSA定位的算法,最早在2013年被提出(Takagi)。它的基本原理是,构建子代分离群体,经过挑选极端性状构建混池后对SNP进行检测,对各混池进行等位基因频率分析,并与其中一个亲本进行比较。与此亲本不同的基因型所占的比例,即为该位点的SNP-index。从下图可以看到,两个位点的SNP-index分别为0.4和1。值得注意的是,这里的reference指的并不是我们进行重测序变异检测的参考基因组,而是我们构建群体所使用的亲本。这也是为什么进行SNP-index计算必须依赖于亲本测序数据的缘故。
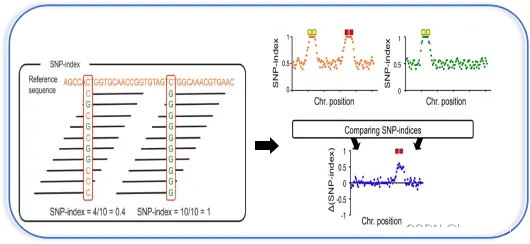
每个混池都得到一组SNP-index数据之后,两个混池相减(上图右),即得到了△SNP-index的结果,代表的是两个混池之间SNP基因型频率的差异。理论上说,不与性状相关的位点,△SNP-index的值应当在0左右,代表混池之间不存在差异;而QTL及其相连锁位置的SNP,△SNP-index值应当呈现较高的数值。△SNP index这种分析方法会存在因统计偏差造成的假阳性位点,这时我们可以通过计算滑窗内所有SNP的△SNP-index,来消除其影响,得到真正QTL所在的基因组区域。
2、 欧几里得距离(ED)
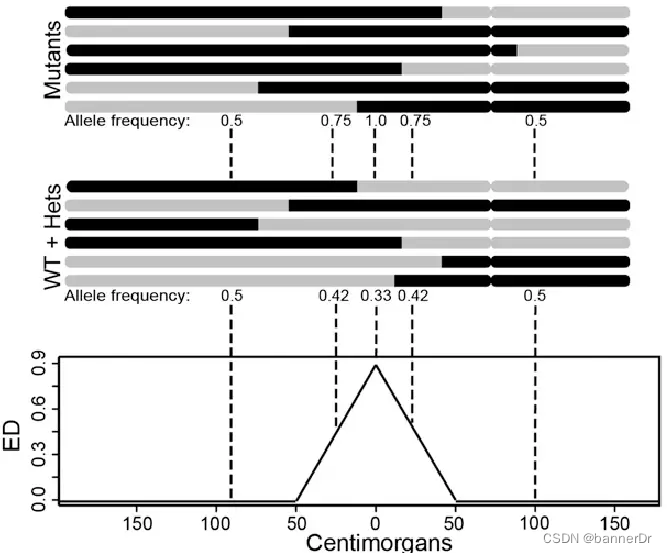
随着BSA技术的发展,SNP-index显示出了一定的局限性,比如亲本数据缺失,林木类较难构建分析群体,ED值的分析方法应运而生。在BSA和BSR中,欧几里得距离可以计算同一个位点上,两个混池之间的等位基因频率。两个极端性状子代混池只在控制性状的QTL及其连锁位点出现差异,所以通过各个位点欧几里得距离的计算,我们可以判断哪些位点更可能是控制对应性状的QTL。计算公式如下:
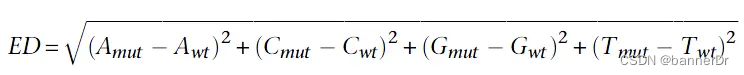
实际应用中,我们在BSA的两组混池之间可能会得到数十万甚至上百万个SNP,有的SNP可能实际与性状无关,但因为抽样偏差,导致计算得到的ED值很高,为了排除统计异常值,我们通常会采用滑窗对在一个窗口内所有位点的ED值进行拟合,消除抽样偏差产生的假阳性结果。而在BSA定位区间计算过程中,会对ED值采取乘方处理,放大ED值的差异,使定位区间更加明显。
3、 Gradedpool-seq(Ridit检验)
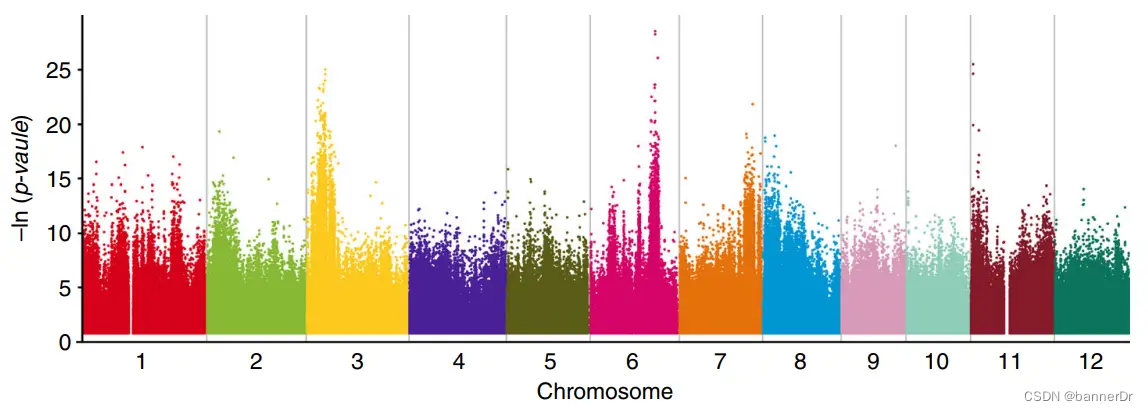
Gradedpool-seq的概念在2019年由韩斌和黄学辉课题组提出并发表于Nature Communication(Wang et al., 2019)。这种方法与常规BSA类似的是,它也是基于性状分离群体中按照性状选择子代个体构成混池(通常加上亲本)进行测序,并进行QTL定位的方式。Ridit是relative to an identified distribution unit一词的缩写,它是一种非参数检验分析方法,用于按等级分组资料的比较。而对于多个混池测序数据,Ridit检验会对每个位点的等位基因频率进行计算,判断其是否显著偏离标准分布,得到一个p值。换言之,这个位点的p值越小,即代表这个位点与性状相关联的可能性越高(与GWAS关联方法类似)。
由于在BSA项目中Ridit检验的对象只有2-4个混池,基因型数据较少,所以当Ridit检验的结果用曼哈顿图的形式展现出来,其噪音非常强烈,很难从中直观地判断我们的候选区间的位置。研究者们选取一定大小的窗口,并且将窗口内的SNP位点进行统计,计算p值低于阈值的位点所占的比例。一般经过这种