DTAComputers_in_Biology_and_Medicine__0">ColdDTA发表在Computers in Biology and Medicine 的一篇一区文章
突出
•
数据增强和基于注意力的特征融合用于药物靶点结合亲和力预测。
•
与其他方法相比,它在 Davis、KIBA 和 BindingDB 数据集上显示出竞争性能。
•
可视化模型权重可以获得可解释的见解。
文章目录
前言
提示:这里可以添加本文要记录的大概内容:
例如:随着人工智能的不断发展,机器学习这门技术也越来越重要,很多人都开启了学习机器学习,本文就介绍了机器学习的基础内容。
提示:以下是本篇文章正文内容,下面案例可供参考
一、摘要
药物靶点亲和力(DTA)的准确预测在药物发现和开发中起着至关重要的作用。最近,深度学习方法在随机拆分的公共数据集上显示出优异的预测性能。然而,仍然需要对这种拆分方法进行验证,以反映实际应用中的实际问题。在冷启动实验设置中,测试集中的药物或蛋白质不会出现在训练集中,深度学习模型的性能通常会显着下降。这表明提高模型的泛化能力仍然是一个挑战。为此,在本研究中,我们提出了ColdDTA:利用数据增强和基于注意力的特征融合来提高预测药物-靶标结合亲和力的泛化能力。具体来说,ColdDTA通过去除药物的子图来生成新的药物-靶标对。基于注意力的特征融合模块也用于更好地捕捉药物-靶点的相互作用。我们在三个基准数据集上进行了冷启动实验,Davis 和 KIBA 数据集上的一致性指数 (CI) 和均方误差 (MSE) 结果表明,ColdDTA 优于五种最先进的基线方法。同时,BindingDB数据集上的受试者工作特征下面积(ROC-AUC)结果表明,ColdDTA在分类任务上也具有更好的性能。此外,可视化模型权重可以获得可解释的见解。总体而言,ColdDTA可以较好地解决现实的DTA预测问题。该代码已向公众开放。
二、引言
药物开发是一个漫长而昂贵的过程。根据调查,开发一种新药大约需要13年时间,成本为0.6-26亿美元,成功率不到10%[1]。这促使人们使用计算方法来降低成本和提高效率。药物分子与具有适当化学特性和亲和力的靶标结合的过程是治愈疾病的关键标准之一。因此,准确预测药物-靶标结合亲和力对于药物发现至关重要[2]。
使用计算机进行药物靶标亲和力(DTA)预测的方法可分为基于结构的方法、基于配体的方法和混合方法[3]。分子对接是最早和使用最广泛的基于结构的方法之一。然而,这种方法需要蛋白质的3D结构和较长的计算时间,即使是现在,仍然有大量未知的蛋白质结构。尽管AlphaFold2能够预测大多数蛋白质[4],但并非所有结果都非常准确[5]。基于配体的方法假设具有相似化学性质的药物分子具有相同的化学活性,因此可以与相似的靶标相互作用。然而,当已知配体数量不足时,基于配体的方法的预测结果往往变得不可靠。混合方法有望克服上述局限性。其中之一,传统的机器学习方法,如支持向量机(SVM)和随机森林(RF),需要特征工程来提取药物和蛋白质特征[6],特征工程非常耗时,并且会因信息丢失而影响预测性能。近年来,已经开发了几种深度学习方法,如DeepDTA [7]和GraphDTA [8],用于药物-靶标相互作用预测。
一些深度学习模型使用基于序列的输入数据,如WideDTA [9],用简化分子输入线输入系统(SMILES)表示药物,用氨基酸序列表示蛋白质,然后通过卷积神经网络(CNN)或递归神经网络提取特征(RNN)。
但这种方法缺乏分子拓扑结构。
为了弥补这一不足,许多方法将药物视为图结构,化合物的原子和化学键对应于图的顶点和边缘,然后通过图神经网络(GNN)进行特征提取。DeepGS [10]、MolTrans [11]和MgraphDTA [12]都以图表示药物,并在一些公共数据集上表现出优异的预测性能。同样,一些研究也关注蛋白质在药物靶标结合过程中的作用,其中 DrugVQA [13]、AttentionSiteDTI [14] 和 GSAML-DTA [15] 使用接触图来表示蛋白质,实验结果表明,使用蛋白质的 3D 结构可以做出更好的预测。Transformer [16] 也被用于预测药物-靶标相互作用。Chen等人提出的TransformerCPI模型将药物-靶标相互作用视为二元分类问题,并使用编码器-解码器架构进行模型训练,最终预测两者是否可以相互作用[17]。
一些深度学习模型使用基于序列的输入数据,如WideDTA [9],用简化分子输入线输入系统(SMILES)表示药物,用氨基酸序列表示蛋白质,然后通过卷积神经网络(CNN)或递归神经网络提取特征(RNN)。但这种方法缺乏分子拓扑结构。为了弥补这一不足,许多方法将药物视为图结构,化合物的原子和化学键对应于图的顶点和边缘,然后通过图神经网络(GNN)进行特征提取。DeepGS [10]、MolTrans [11]和MgraphDTA [12]都以图表示药物,并在一些公共数据集上表现出优异的预测性能。同样,一些研究也关注蛋白质在药物靶标结合过程中的作用,其中 DrugVQA [13]、AttentionSiteDTI [14] 和 GSAML-DTA [15] 使用接触图来表示蛋白质,实验结果表明,使用蛋白质的 3D 结构可以做出更好的预测。Transformer [16] 也被用于预测药物-靶标相互作用。Chen等人提出的TransformerCPI模型将药物-靶标相互作用视为二元分类问题,并使用编码器-解码器架构进行模型训练,最终预测两者是否可以相互作用[17]。
尽管一些深度学习方法在DTA问题上表现出出色的预测性能,但这些方法大多是在随机拆分的数据集上进行评估的(测试集中的药物和目标已经出现在训练集中),这可能导致信息泄露,使结果过于乐观[18]。从应用的角度来看,大多数蛋白质或药物不会出现在训练集中[19]。当数据集根据更现实的场景进行拆分时,许多模型的预测性能急剧下降[20],有时预测性能比传统机器学习方法更差[21]。这表明当前DTA模型的泛化能力仍有待提高。
为了提高模型的泛化能力,使其在真实场景中发挥更大的作用,我们首先考虑了数据增强方法的运用。数据增强方法已广泛应用于计算机视觉[[22],[23],[24]]和自然语言处理[[25],[26],[27]],并已被证明可以有效提高模型泛化。还提出了多种有效的数据增强方法来预测分子特性[28,29]。然而,据我们所知,数据增强尚未用于提高使用分子图表示药物的 DTA 预测方法的泛化性能。这可能是由于生物化学告诉我们,化合物中原子的微小变化也可能导致分子物理和化学性质的显着变化,以及小药物分子与靶标之间结合亲和力的变化。如图1所示,对于邻苯二酚分子,去除一些原子为苯酚会将其水溶性从溶于水变为微溶于水。另一方面,特征融合是指来自不同层或分支的特征的组合,在现代网络架构中无处不在,使用合理的特征融合方法可以有效提高模型性能。但现有的大多数DTA预测方法只是简单地将药物和蛋白质的特征串联起来,进行最终结果预测,这限制了药物与靶点之间的相互作用,限制了模型的预测和泛化性能。
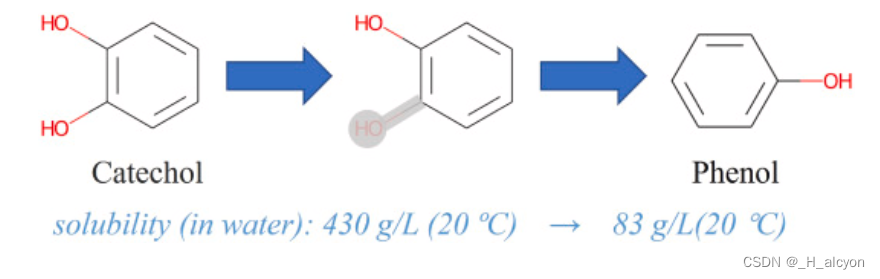
图 1.影响其性质的原子变化的图示。
在这项工作中,我们提出了一种称为ColdDTA的药物靶标亲和力训练方法,该方法使用数据增强和基于注意力的特征融合来预测药物和蛋白质之间的结合亲和力。具体来说,在数据处理阶段,将一定比例的药物子图移除,并与原始目标构成新的数据对,然后在训练阶段使用。此外,ColdDTA使用药物-蛋白质特征融合模块来取代简单的特征串联。通过对两个基准数据集进行实验,我们发现与其他方法相比,ColdDTA的整体性能最佳。我们的消融实验结果也证明了数据增强方法和药物-靶点融合模块的有效性。本研究的主要贡献总结如下:
1.
采用一种新的数据增强方法,通过去除药物的子图,与原目标形成新的数据对,可以有效提高模型的泛化能力。
该文提出一种基于注意力的药物与靶点数据融合方法,该方法能够更好地将药物特征与蛋白质特征融合,有利于模型进行亲和值预测。
在两个公开数据集上进行了大量实验,验证了在接近真实实验环境的数据集设置下,ColdDTA的预测能力与基线模型相比有显著提高。此外,实验还证明了数据增强和药物靶点融合的有效性。
三、方法
1.概述
我们提出了一种称为ColdDTA的端到端训练方法来提高DTA预测性能。我们对 DTA 问题的定义如下:让
是一批标记的数据,其中
是小分子药物的SMILES表示,
是蛋白质,并且
是通过实验获得的结合亲和力值。模型的整体训练过程如图 2 所示。它由数据增强模块、药物特征提取模块、蛋白质特征提取模块、药物-靶点融合模块和预测模块五部分组成。
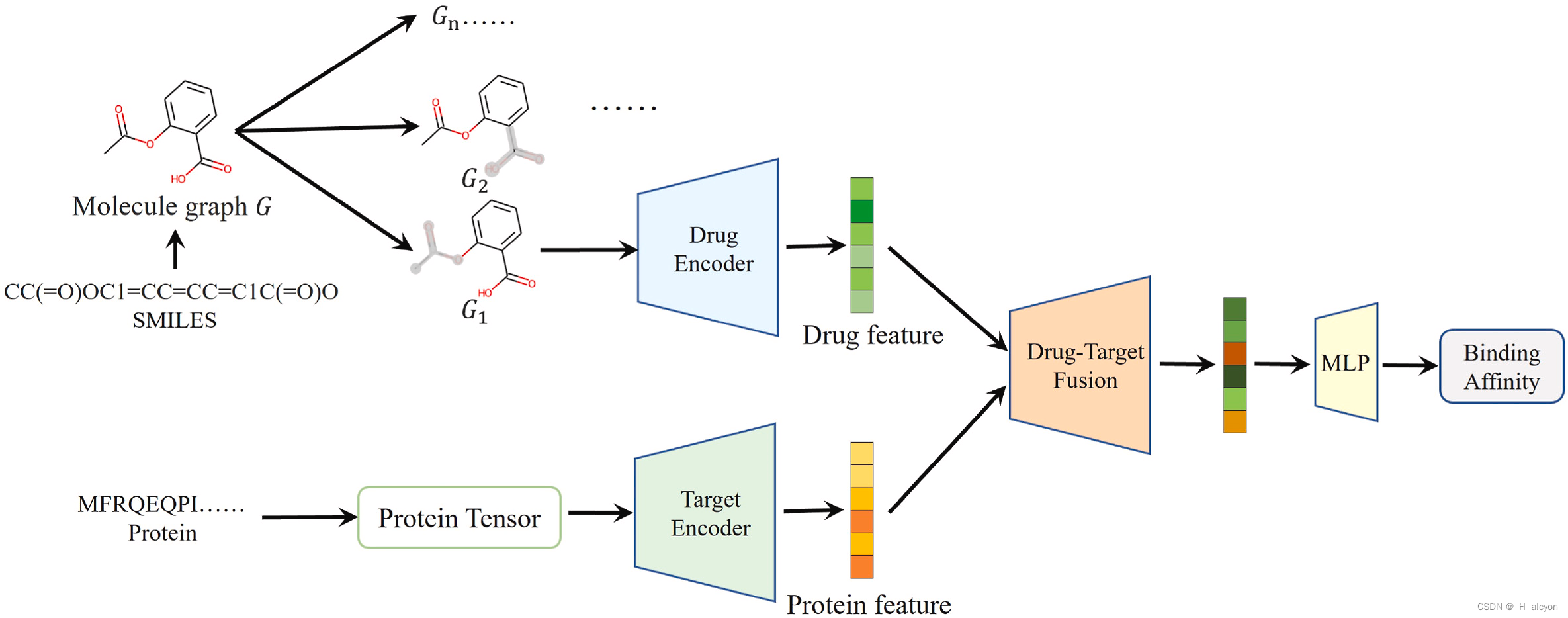
图 2.拟议的 ColdDTA 的框架。首先,去除药物的子图,并与原始蛋白质形成新的对。其次,利用GNN和CNN分别提取分子图和蛋白质的特征;第三,将上一步的特征输入到药物-靶点融合模块进行交互,以获得更有效的特征表示。最后,将融合特征输入MLP以预测结合亲和力。
2.数据增强策略
数据增强是提高神经网络数据效率、增强泛化性能的必要条件[30]。使用数据增强方法可以提高模型的预测性能似乎很明显,但很少有研究使用这种策略来预测药物-靶点相互作用预测问题,因此我们在DTA预测中引入了一种新的数据增强策略。具体来说,在药物-靶点配对中,我们首先随机选择分子图中的一个原子作为起始节点,然后去除起始节点并递归地去除其相邻节点,直到去除一定比例的子图,而相应的靶点和结合亲和力保持不变。在这个过程中,原子基团的去除是完全随机的,这类似于分子性质预测的对比学习[31]。图 3 显示了数据增强过程的输出。在训练阶段,一对药物化合物和靶蛋白通过去除子图产生多个新对。需要注意的是,在测试阶段不会删除任何子图。
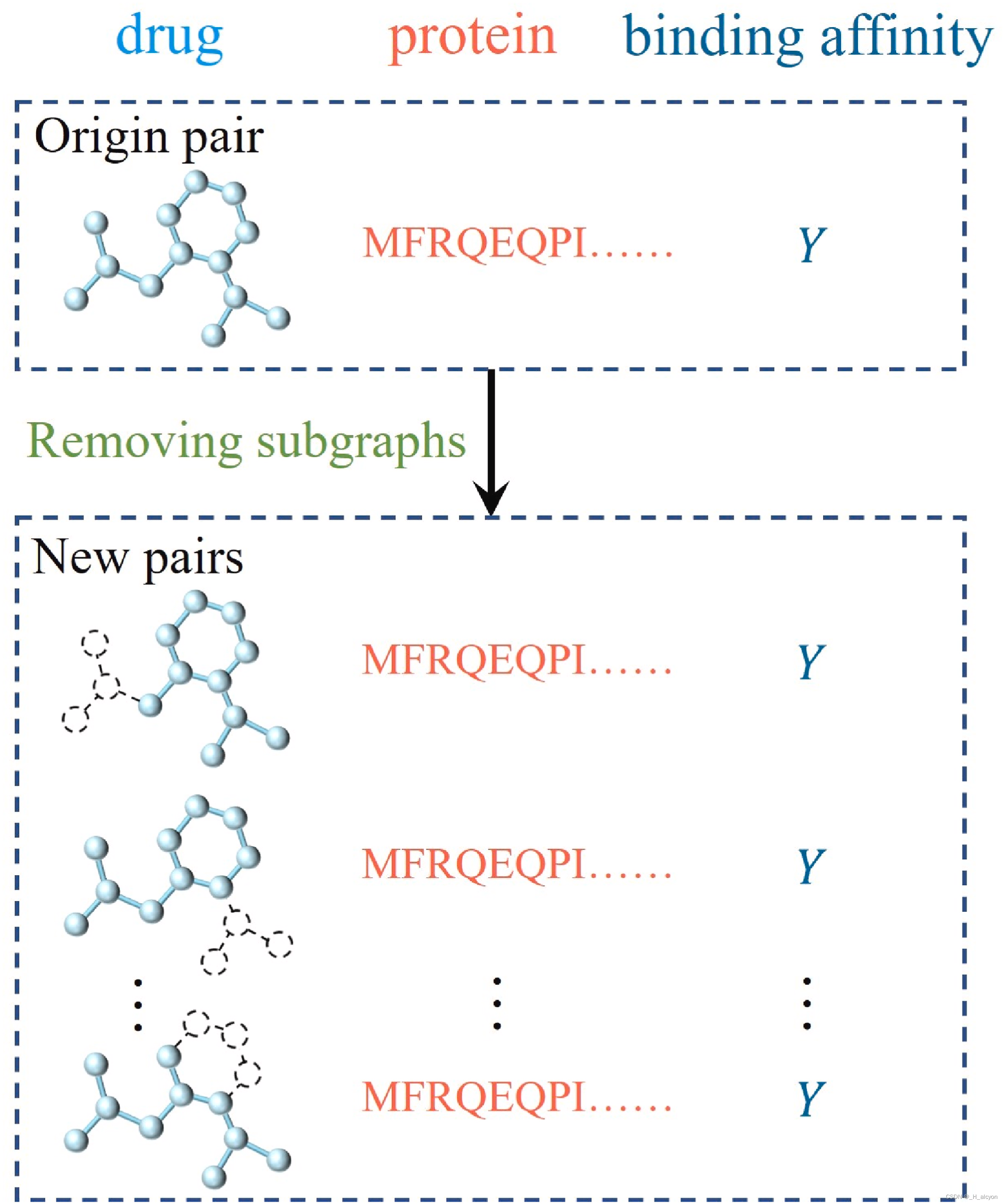
药物-靶点相互作用(DTI)预测是利用计算方法和模型来预测药物与生物靶点之间的相互作用。这一领域可以帮助研究人员了解药物的作用机制、发现新的药物靶点以及筛选候选药物。近年来,许多融合深度药物和靶点相互作用特征的DTI模型出现,弥补了浅层药物和靶点特征信息不完整的困境¹。
在药物-靶点配对中,数据增强是一种常用的技术,旨在提高模型的性能。具体来说,数据增强过程通常包括以下步骤:
- 起始节点选择:从药物分子图中随机选择一个原子作为起始节点。
- 递归去除子图:去除起始节点并递归地去除其相邻节点,直到去除一定比例的子图。
- 保持靶点和结合亲和力不变:在去除子图的过程中,保持相应的靶点和结合亲和力不变。
- 随机原子基团去除:原子基团的去除是完全随机的,类似于分子性质预测的对比学习。
- 训练阶段生成新对:在训练阶段,一对药物化合物和靶蛋白通过去除子图产生多个新对。
- 测试阶段不删除子图:需要注意的是,在测试阶段不会删除任何子图。
这样的数据增强方法有助于提高模型的泛化能力,使其在测试集上表现更好。¹²
如果你对药物-靶点相互作用预测还有其他问题,欢迎继续提问!
Source: Conversation with Bing, 04/05/2024
(1) 论文笔记38|Flexible drug-target interaction prediction with interactive … https://zhuanlan.zhihu.com/p/689761796.
(2) ColdDTA:利用数据增强和基于注意力的特征融合进行药物靶标结合亲和力预测-CSDN博客. https://blog.csdn.net/weixin_46042619/article/details/138422638.
(3) 打破药物靶点亲和力预测中数据稀缺的障碍。,Briefings in Bioinformatics - X-MOL. https://www.x-mol.com/paper/1729938709820493824/t.
(4) AI再发力?基于结构的高精度对接,如何致力更精准药物设计?. https://www.drugtimes.cn/2023/04/11/aizaifalijiyujiegoudegaojingduduijieruhezhiligengjingzhun/.
在这篇文章中,分子性质预测的对比学习是一种数据增强策略,用于提高模型在药物靶点亲和力(DTA)预测中的泛化能力。具体来说,这种策略通过随机去除药物分子图中的原子基团来生成新的药物-靶标对,从而增加训练数据的多样性。这类似于计算机视觉中的对比学习,其中通过对图像进行随机变换来创建正负样本对,以帮助模型学习区分不同的特征。在这里,通过去除原子基团,模型可以学习到更多关于药物分子结构和功能的信息,这有助于提高其在新药物或靶标上的预测准确性。
3.药物特征提取模块

在图卷积网络(GCN)中,每个原子的特征向量通常是通过考虑原子类型、原子杂化模式、氢原子总数和原子价等属性来计算的。这些特征向量可以帮助模型理解分子的化学性质。下面是一个简化的例子,展示了如何计算一个原子的特征向量:
-
原子类型:为每种原子类型分配一个独热编码(one-hot encoding)。例如,如果我们只考虑碳(C)、氮(N)、氧(O)和氢(H),那么碳可以表示为 [ 1 , 0 , 0 , 0 ] [1, 0, 0, 0] [1,0,0,0],氮为 [ 0 , 1 , 0 , 0 ] [0, 1, 0, 0] [0,1,0,0],依此类推。
-
原子杂化模式:同样使用独热编码来表示。例如,sp3杂化可以表示为 [ 1 , 0 , 0 ] [1, 0, 0] [1,0,0],sp2为 [ 0 , 1 , 0 ] [0, 1, 0] [0,1,0],sp为 [ 0 , 0 , 1 ] [0, 0, 1] [0,0,1]。
-
氢原子总数和原子价:这些可以直接用数值表示,例如,一个碳原子可能有4个氢原子,原子价为4。
将这些特征组合起来,就可以得到一个原子的特征向量。例如,一个sp3杂化的碳原子,有4个氢原子,原子价为4,其特征向量可以是 [ 1 , 0 , 0 , 0 , 1 , 0 , 0 , 4 , 4 ] [1, 0, 0, 0, 1, 0, 0, 4, 4] [1,0,0,0,1,0,0,4,4]。
在图卷积网络中,每个顶点(原子)的特征不仅包括其自身的特征,还包括其相邻顶点(原子)的特征。通过多轮的特征聚合,每个顶点可以收集到越来越广泛的邻域信息。这通常是通过以下步骤实现的:
-
聚合:对于每个顶点,收集其所有邻居的特征,并将它们聚合成一个单一的向量。这可以通过求和、平均或其他聚合函数来完成。
-
更新:将聚合后的邻居特征与顶点自身的特征结合起来,通常是通过一个神经网络层来实现,以生成新的顶点特征。
-
传播:重复上述过程多轮,每一轮都会更新顶点的特征,使其包含更远距离邻居的信息。
通过这种方式,模型可以捕捉到分子结构中的局部和全局信息,从而更好地预测分子的性质。这种方法在药物发现和材料科学中特别有用,因为它允许模型学习复杂的分子结构-性质关系。
当然可以。在图神经网络(GNN)的上下文中,公式(1)通常表示为一个更新规则,用于在每次迭代中更新图中顶点的特征向量。这里是公式(1)的一般形式及其组成部分的解释:
h i ( k + 1 ) = U k ( h i ( k ) , ∑ j ∈ N ( i ) h j ( k ) ) h_i^{(k+1)} = U_k \left( h_i^{(k)}, \sum_{j \in N(i)} h_j^{(k)} \right) hi(k+1)=Uk hi(k),j∈N(i)∑hj(k)
-
( h_i^{(k+1)} ): 这是顶点i在第k+1轮迭代后的特征向量。它是模型试图学习的新特征,包含了顶点自身以及其邻居的信息。
-
( U_k ): 这是一个更新函数,通常是一个神经网络层,如全连接层或更复杂的结构。它负责将顶点的当前特征和其邻居的特征结合起来,生成新的特征向量。
-
( h_i^{(k)} ): 这是顶点i在第k轮迭代的特征向量。它代表了顶点在当前迭代之前的状态。
-
( \sum_{j \in N(i)} h_j^{(k)} ): 这是顶点i所有邻居顶点在第k轮迭代的特征向量之和。这个求和过程是特征聚合的一部分,它允许顶点收集其邻居的信息。
在每次迭代中,每个顶点都会通过聚合邻居的特征并结合自身的特征来更新其特征向量。这个过程允许GNN捕捉到图中顶点的局部连接模式,从而学习到更丰富的顶点表示。这种特征的更新和传播对于理解复杂的图结构,如分子结构,以及预测其性质,如药物-靶标结合亲和力等任务至关重要。
在图神经网络(GNN)中,公式 ( (1+w)h_i + h_j ) 的集合表示顶点 ( i ) 的特征向量 ( h_i ) 与其邻居 ( j ) 的特征向量 ( h_j ) 之间的关系。这里的加法操作是为了聚合顶点 ( i ) 自身的信息和其邻居的信息。这种聚合有助于模型捕捉到图中的局部结构信息,从而更好地理解分子的性质或社交网络中的关系模式。
-
为什么要相加:
- 聚合邻居信息:通过将顶点 ( i ) 的特征与其所有邻居的特征相加,我们可以聚合邻域信息,这对于理解顶点在图中的角色至关重要。
- 捕捉局部结构:加法操作使得每个顶点的特征不仅包含自身的信息,还包含其邻居的信息,这有助于模型学习到顶点的局部连接模式。
-
( w ) 的值是怎么来的:
- 可学习的参数:( w ) 是一个权重参数,它在模型训练过程中通过反向传播算法学习得到。它的作用是调整顶点自身特征在特征更新过程中的重要性。
- 优化目标:( w ) 的值是为了最小化模型在特定任务上的损失函数,例如分类误差或回归误差,从而使模型能够更准确地预测。
在实际应用中,( w ) 和 ( h ) 的具体值将通过模型训练确定,通常是使用梯度下降或其他优化算法来调整这些参数,以便模型能够在给定的任务上表现最好。
让我们通过一个具体的例子来看看如何在图神经网络(GNN)中更新顶点的特征向量。
假设我们有一个小分子,它由三个原子组成:一个碳原子(C),一个氧原子(O),和一个氢原子(H)。我们将使用一个简化的图卷积网络来更新这些原子的特征向量。
-
初始特征向量:
- 碳原子(C): h C ( 0 ) = [ 1 , 0 , 0 , 0 , 4 ] h_C^{(0)} = [1, 0, 0, 0, 4] hC(0)=[1,0,0,0,4]
- 氧原子(O): h O ( 0 ) = [ 0 , 1 , 0 , 2 , 6 ] h_O^{(0)} = [0, 1, 0, 2, 6] hO(0)=[0,1,0,2,6]
- 氢原子(H): h H ( 0 ) = [ 0 , 0 , 1 , 1 , 1 ] h_H^{(0)} = [0, 0, 1, 1, 1] hH(0)=[0,0,1,1,1]
这里,我们假设特征向量包含原子类型的独热编码和原子价。
-
邻居特征聚合:
- 假设碳原子(C)和氧原子(O)相连,氢原子(H)和碳原子(C)相连。
- 在第一轮迭代中,我们聚合每个原子的邻居特征向量。
-
特征更新:
- 使用一个简单的更新函数,例如元素加和,来更新每个原子的特征向量。
例如,对于碳原子(C),其更新后的特征向量可以通过以下方式计算:
h C ( 1 ) = h C ( 0 ) + ∑ j ∈ N ( C ) h j ( 0 ) h_C^{(1)} = h_C^{(0)} + \sum_{j \in N(C)} h_j^{(0)} hC(1)=hC(0)+j∈N(C)∑hj(0)
这里,( N© ) 表示碳原子的邻居集合,即氧原子(O)和氢原子(H)。因此,我们有:
h C ( 1 ) = [ 1 , 0 , 0 , 0 , 4 ] + [ 0 , 1 , 0 , 2 , 6 ] + [ 0 , 0 , 1 , 1 , 1 ] h_C^{(1)} = [1, 0, 0, 0, 4] + [0, 1, 0, 2, 6] + [0, 0, 1, 1, 1] hC(1)=[1,0,0,0,4]+[0,1,0,2,6]+[0,0,1,1,1]
h C ( 1 ) = [ 1 , 1 , 1 , 3 , 11 ] h_C^{(1)} = [1, 1, 1, 3, 11] hC(1)=[1,1,1,3,11]
这个新的特征向量 ( h_C^{(1)} ) 现在包含了碳原子自身的信息以及其邻居的信息。通过这种方式,GNN能够在每次迭代中捕捉到更多的局部结构信息。
请注意,这只是一个非常简化的例子,实际的GNN模型会使用更复杂的特征和更新函数。此外,为了提高模型的性能,通常会使用非线性激活函数和可学习的参数。如果您需要更详细的解释或有其他问题,请告诉我!
公式(2)描述了如何计算整个分子图的特征向量。这里, y G \mathbf{y}_G yG 是分子图的特征向量, h v ( k ) \mathbf{h}_v(k) hv(k) 是时间步长 k k k 时顶点 v v v 的特征向量, ∣ V ∣ |V| ∣V∣ 是分子图中顶点的数量。公式可以表示为:
y G = 1 ∣ V ∣ ∑ v ∈ V h v ( k ) \mathbf{y}_G = \frac{1}{|V|} \sum_{v \in V} \mathbf{h}_v(k) yG=∣V∣1v∈V∑hv(k)
这个公式通过对所有顶点的特征向量进行平均,来得到整个图的特征向量。这是图神经网络中常用的一个步骤,用于将图中所有节点的信息汇总成一个全局的图表示。
4.蛋白质特征提取模块

5.药物-靶点融合模块

在当前页面的部分2.4 "蛋白质特征提取模块"中,公式(3)描述了如何通过一维卷积层(CNN)从蛋白质序列中提取特征。这里是公式的详细解释:
-
公式:
X p ( l + 1 ) = B ∘ δ ∘ F ( X p ( l ) ) \mathbf{X}_p^{(l+1)} = \mathbf{B} \circ \delta \circ \mathbf{F}(\mathbf{X}_p^{(l)}) Xp(l+1)=B∘δ∘F(Xp(l)) -
变量解释:
- ( \mathbf{X}_p^{(l)} ):第 ( l ) 层的隐藏蛋白质表示。[1][1]
- ( \mathbf{X}_p^{(0)} ):初始输入矩阵,即氨基酸序列的嵌入表示。
- ( \mathbf{F} ):CNN层,用于提取序列特征。
- ( \delta ):ReLU激活函数,增加非线性。
- ( \mathbf{B} ):批量归一化(BN),用于调整输入分布,加速训练。[2][2]
-
操作解释:
- ( \circ ):函数组合操作,表示将一个函数的输出作为另一个函数的输入。
- 公式中,首先通过CNN层 ( \mathbf{F} ) 提取特征,然后应用ReLU激活函数 ( \delta ) 增加非线性,最后进行批量归一化 ( \mathbf{B} )。
这个过程在每个卷积层重复,以提取蛋白质序列的全局特征,为后续的药物-靶点结合亲和力预测提供信息。
图卷积网络(GCN)处理分子之间的连接关系的方式是通过考虑分子中原子(顶点)之间的化学键(边)。在GCN中,每个原子由一个特征向量表示,而原子之间的化学键则通过图中的边来表示。这些边决定了特征向量如何在网络中传播和更新。下面是GCN处理分子连接关系的基本步骤:
-
初始化特征向量:每个原子的初始特征向量通常包含原子类型、原子价、杂化类型等信息。
-
边的表示:分子中的每个化学键都表示为图中的一条边。这些边不仅表示原子之间的连接,还可以包含边的类型,如单键、双键或三键。
-
特征传播:在每次迭代中,原子的特征向量会根据其邻居(通过化学键连接的其他原子)的特征进行更新。这个过程通常涉及到特征的聚合和组合。
-
聚合函数:定义一个聚合函数来收集邻居的特征。这个函数可以是简单的求和、平均或更复杂的操作,如加权求和,其中权重可以表示化学键的类型或强度。
-
更新规则:定义一个更新规则来结合原子自身的特征和聚合的邻居特征,生成新的特征向量。这个规则通常是一个神经网络层,如全连接层。
-
非线性激活:在每次特征更新后,通常会应用一个非线性激活函数,如ReLU,以增加模型的表达能力。
通过这种方式,GCN能够学习到分子结构的复杂模式,并用于预测分子的性质,如溶解度、毒性或药物活性。这种方法在药物发现和材料科学中特别有用,因为它允许模型学习复杂的分子结构-性质关系。
如果您需要更多具体的例子或有其他问题,请随时告诉我!
20240503
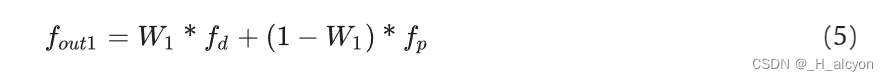
公式(5)描述了药物-靶点融合模块中的第一次特征融合过程。这个过程使用了注意力权重 ( \mathbf{W}_1 ) 来结合药物特征 ( \mathbf{f}_d ) 和蛋白质特征 ( \mathbf{f}p ),生成融合后的特征 ( \mathbf{f}{\text{out1}} )。具体来说,公式如下:
f out1 = W 1 ∗ f d + ( 1 − W 1 ) ∗ f p \mathbf{f}_{\text{out1}} = \mathbf{W}_1 * \mathbf{f}_d + (1 - \mathbf{W}_1) * \mathbf{f}_p fout1=W1∗fd+(1−W1)∗fp
其中,( \mathbf{W}_1 ) 是通过另一个公式计算得到的注意力权重,它决定了在融合特征中药物和蛋白质各自所占的比重。这个过程的目的是为了更好地捕捉药物和蛋白质之间的相互作用信息,从而提高模型在药物-靶点结合亲和力预测任务中的性能和泛化能力。[1][1]
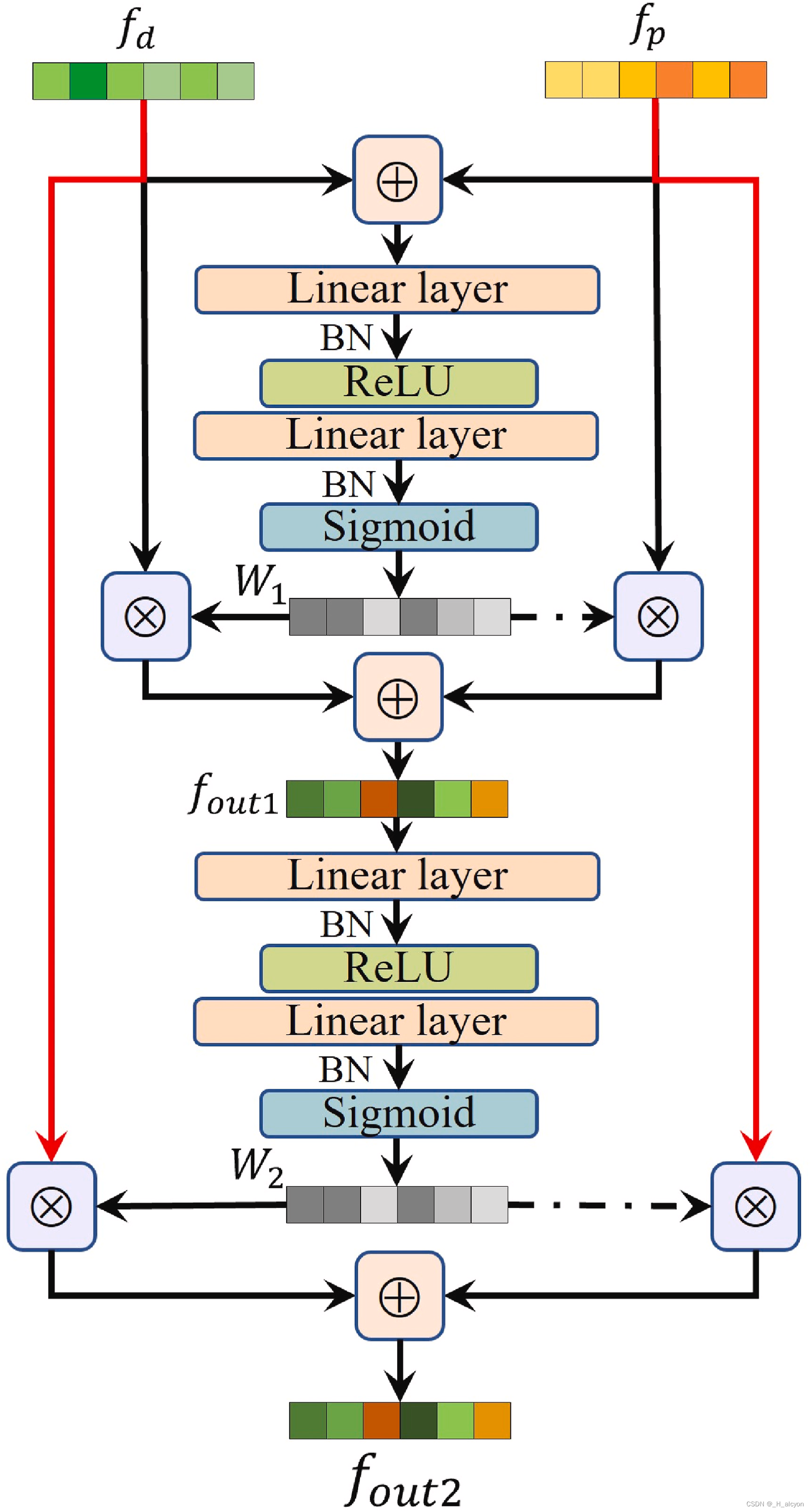
图 4.拟议的药物-靶点融合的图示。
图4展示了药物-靶点融合模块的工作原理,该模块基于注意力机制来更好地捕捉药物与蛋白质之间的相互作用。[1][1][2][2]以下是该模块的具体操作步骤和运算符号解释:
-
输入特征:给定药物特征 f d ∈ R d f_d \in \mathbb{R}^d fd∈Rd 和蛋白质特征 f p ∈ R d f_p \in \mathbb{R}^d fp∈Rd,它们具有相同的维度 d d d。
-
注意力权重计算:首先计算注意力权重 W 1 ∈ R d W_1 \in \mathbb{R}^d W1∈Rd,使用以下公式:
W 1 = σ ∘ B ∘ θ ∘ δ ∘ B ∘ θ ( f d ⊕ f p ) W_1 = \sigma \circ B \circ \theta \circ \delta \circ B \circ \theta (f_d \oplus f_p) W1=σ∘B∘θ∘δ∘B∘θ(fd⊕fp)
其中, ⊕ \oplus ⊕ 表示元素加法, ∘ \circ ∘ 表示函数组合, θ \theta θ 表示线性层, δ \delta δ 表示ReLU激活函数, B B B 表示批量归一化, σ \sigma σ 表示Sigmoid函数。 -
特征融合:使用注意力权重进行第一次特征融合,得到新的特征 f o u t 1 ∈ R d f_{out1} \in \mathbb{R}^d fout1∈Rd:
f o u t 1 = W 1 ∗ f d + ( 1 − W 1 ) ∗ f p f_{out1} = W_1 * f_d + (1 - W_1) * f_p fout1=W1∗fd+(1−W1)∗fp -
二次注意力权重计算:为了提高融合特征的质量,再次使用注意力模块计算新的融合权重 W 2 ∈ R d W_2 \in \mathbb{R}^d W2∈Rd:
W 2 = σ ∘ B ∘ θ ∘ δ ∘ B ∘ θ ( f o u t 1 ) W_2 = \sigma \circ B \circ \theta \circ \delta \circ B \circ \theta (f_{out1}) W2=σ∘B∘θ∘δ∘B∘θ(fout1) -
最终融合输出:使用新的注意力权重 W 2 W_2 W2 进行最终融合,得到最终输出特征 f o u t 2 ∈ R d f_{out2} \in \mathbb{R}^d fout2∈Rd:
f o u t 2 = W 2 ∗ f d + ( 1 − W 2 ) ∗ f p f_{out2} = W_2 * f_d + (1 - W_2) * f_p fout2=W2∗fd+(1−W2)∗fp
这个基于注意力的特征融合方法有效地捕捉了药物-靶点相互作用的信息,从而提高了DTA预测的准确性和模型的泛化能力。[1][1]**

6.预测模块

当然可以。在2.6节中描述的预测模块是药物-靶点亲和力(DTA)预测任务的核心部分。这个模块通常使用前面提取和融合的特征来预测药物分子与蛋白质靶点之间的结合亲和力。以下是预测模块的详细解释:
-
特征输入:模块接收来自药物-靶点融合模块的融合特征向量,这些特征向量包含了药物分子和蛋白质靶点的重要信息。
-
全连接层:融合特征首先通过一个或多个全连接层(也称为密集层),这些层可以进一步处理特征,提取更高级的表示。
-
激活函数:全连接层后通常会有非线性激活函数,如ReLU或Sigmoid,它们增加模型的非线性,使模型能够捕捉更复杂的关系。
-
输出层:最后,特征通过一个输出层,该层通常只有一个神经元,用于预测亲和力值。对于回归任务,输出层可能不使用激活函数或使用线性激活函数。
-
损失函数:在训练过程中,模型的输出与真实的亲和力值进行比较,计算损失函数(如均方误差),并通过反向传播更新模型的权重。
-
优化算法:使用优化算法(如梯度下降)来最小化损失函数,从而调整模型参数,提高预测的准确性。
整个预测模块的目标是学习药物分子和蛋白质靶点之间的复杂相互作用,并准确预测它们之间的结合亲和力。这对于药物设计和发现过程中识别潜在的药物候选物非常重要。
如果您需要更多具体的信息或有其他问题,请随时告诉我!
交叉熵损失(Cross-Entropy Loss)和均方误差损失(Mean Squared Error, MSE Loss)是两种常用的损失函数,它们在机器学习模型的训练中用于不同的目的:
-
交叉熵损失:
- 用途:通常用于分类问题,特别是二分类或多分类问题。
- 计算:测量实际输出(概率分布)和预测输出(概率分布)之间的差异。
- 公式:对于二分类问题,公式为 L ( y , y ^ ) = − [ y log ( y ^ ) + ( 1 − y ) log ( 1 − y ^ ) ] L(y, \hat{y}) = -[y \log(\hat{y}) + (1 - y) \log(1 - \hat{y})] L(y,y^)=−[ylog(y^)+(1−y)log(1−y^)],其中 ( y ) 是真实标签,( \hat{y} ) 是预测概率。
- 特点:当预测错误时,惩罚(损失)较大,尤其是当预测概率与实际标签相差很大时。
-
均方误差损失:
- 用途:通常用于回归问题,即预测连续值。
- 计算:测量实际输出值和预测输出值之间差异的平方的平均值。
- 公式: L ( y , y ^ ) = 1 n ∑ i = 1 n ( y i − y ^ i ) 2 L(y, \hat{y}) = \frac{1}{n} \sum_{i=1}^{n} (y_i - \hat{y}_i)^2 L(y,y^)=n1i=1∑n(yi−y^i)2,其中 ( y_i ) 是真实值,( \hat{y}_i ) 是预测值,( n ) 是样本数量。
- 特点:对于每个点的误差都给予相同的权重,而且误差的大小会被平方,因此对于较大误差的惩罚更重。
总的来说,选择哪种损失函数取决于具体的任务类型和目标。交叉熵损失更适合处理概率输出的分类任务,而均方误差损失更适合处理数值预测的回归任务。如果您有更多问题或需要进一步的解释,请随时告诉我!
7.数据集


当然可以。这三个参数在药物化学和酶学中非常重要,它们用于描述小分子与蛋白质或酶之间的相互作用强度。
-
解离常数 (Kd): Kd 是指在化合物与其靶标蛋白质结合后,50%的复合物解离成游离状态时对应的化合物浓度。Kd 值越小,表示亲和力越强,即化合物与靶标蛋白质结合得越紧密⁷。
-
抑制常数 (Ki): Ki 是指在酶促反应中,50%的酶被抑制剂结合时对应的游离抑制剂的浓度。Ki 值越小,抑制剂对酶的抑制能力越强⁴。
-
半最大抑制浓度 (IC50): IC50 是指在体外实验中,能够抑制特定生物过程(如酶活性、细胞增殖等)50%所需的抑制剂浓度。IC50 值用于衡量抑制剂的功效,值越小,抑制剂越有效¹。
这些参数在药物设计和评估中非常关键,因为它们可以帮助研究者理解药物与其靶标之间的相互作用,并预测药物的效力。
Source: Conversation with Bing, 04/05/2024
(1) KD 值:抗体亲和力的定量测定 - Abcam中文官网. https://www.abcam.cn/primary-antibodies/kd-value-a-quantitive-measurement-of-antibody-affinity.
(2) IC50、EC50、Ki、Kd、Ka、Km、Kon、Koff傻傻分不清?可能是目前为止最详尽讨论文章 - 知乎. https://zhuanlan.zhihu.com/p/29900912.
(3) IC50 - Wikipedia. https://en.wikipedia.org/wiki/IC50.
(4) 50% of what? How exactly are IC50 and EC50 defined?. https://www.graphpad.com/support/faq/50-of-what-how-exactly-are-ic50-and-ec50-defined/.
(5) IC50 Calculator | AAT Bioquest. https://www.aatbio.com/tools/ic50-calculator.
(6) IC50、pIC50、EC50、ED50、Ki、Kd、KD、Ka、Km、Kon、Koff概念辨析 纽普生物. https://www.novopro.cn/articles/202001111199.html.
(7) 正确理解衡量蛋白质-小分子相互作用的生化指标Kd,Ki以及Km - 知乎. https://zhuanlan.zhihu.com/p/21656428.
DeepPurpose软件包
是一个基于深度学习的分子建模和预测工具包,它支持药物-靶点相互作用预测、化合物性质预测、蛋白质-蛋白质相互作用预测和蛋白质功能预测等任务。在处理冷启动问题时,DeepPurpose可能会采用特定的数据划分策略,以确保模型能够在没有先前交互数据的情况下进行训练和预测。
具体到冷启动划分的实现,DeepPurpose可能会使用以下方法之一或组合:
- 随机划分:将数据集随机划分为训练集、验证集和测试集,确保每个集合中都有新的药物或靶标出现。
- 时间划分:根据药物或靶标的发现时间进行划分,以模拟真实世界中新药物或新靶标的出现。
- 聚类划分:先对药物或靶标进行聚类,然后确保不同的聚类分布在训练集、验证集和测试集中,以此来模拟冷启动场景。
由于DeepPurpose软件包的文档和社区提供的信息可能更加详尽和准确,建议直接查阅其官方GitHub仓库或相关文档¹,以获取具体的实现细节和示例代码。如果您需要进一步的帮助,也可以在相关的技术论坛或社区中寻求支持。如果您有其他问题或需要更多帮助,请告诉我!
Source: Conversation with Bing, 04/05/2024
(1) GitHub - kexinhuang12345/DeepPurpose: A Deep Learning Toolkit for DTI … https://github.com/kexinhuang12345/DeepPurpose.
(2) DeepPurpose · PyPI. https://pypi.org/project/DeepPurpose/.
(3) DeepPurpose. https://deeppurpose.readthedocs.io/_/downloads/en/latest/pdf/.
四、实验
1.评估指标

2.实验设置

DTA_321">3.coldDTA的模型性能
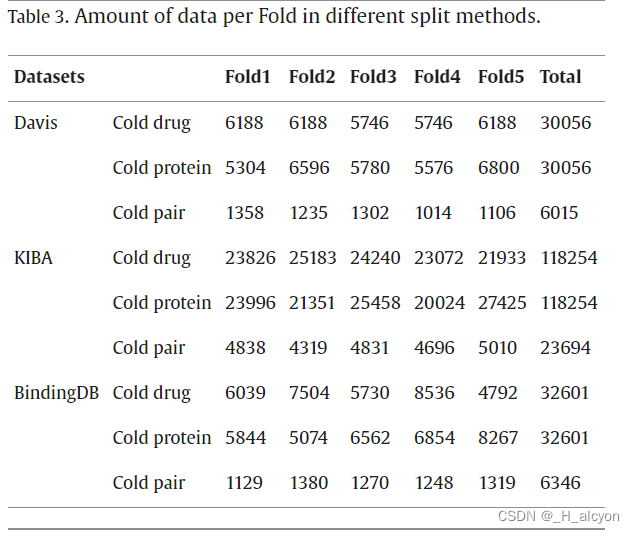
我们将 ColdDTA 的结果与其他预测方法进行了比较,包括传统的机器学习模型:支持向量机 (SVM)、随机森林 (RF) 和深度学习模型:DeepDTA [7]、TransformerCPI [17] 和 MgraphDTA [12]。在一些DTA任务中,RF显示出比深度学习方法更好的性能[41],因此有必要与传统的机器学习方法进行比较。DeepDTA 将 CNN 应用于 SMILES 字符串和蛋白质序列,以提取局部残基模式。当我们将问题转换为分类任务时,在 DeepDTA 的末尾添加了一个 Sigmoid 激活函数,将其转换为二元分类问题,并执行超参数搜索。TransformerCPI 最初是为二元分类任务设计的,为了实现比较,我们替换了它的分类器和损失函数,使其适应回归任务。MgraphDTA构建了一个具有更多图卷积层的超深GNN,以捕获化合物的局部和全局结构,并因此获得了更高的性能。与之前的研究[20]一致,该实验采用了5倍交叉验证方法,其中所有数据被均匀地分为五个部分,其中一部分用作测试集,其余四部分用于训练。这导致了数据集的五种不同方案,平均分数被记录为最终的性能指标。每个折叠中包含的数据量如表3所示。由于冷启动数据拆分方法需要确保测试集中的药物或蛋白质不会出现在训练集中,因此每个折叠中包含的数据量并不完全相等,并且在冷对中,这种拆分方法导致数据总数较少。同时,我们重复了五次实验,每次都选择不同的随机种子。为了保证实验的公正性,所有方法的参数要么采用原始论文,要么通过实验确定,实验在相同的训练集和测试集上进行,但由于数据集拆分过程的随机性,这可能会导致实验结果与其他论文不同。
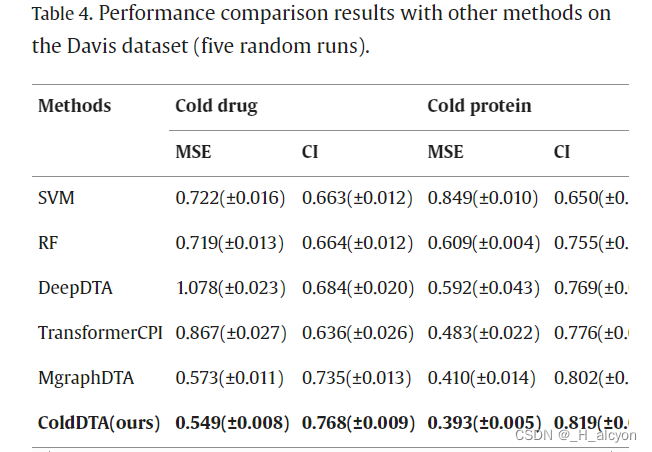
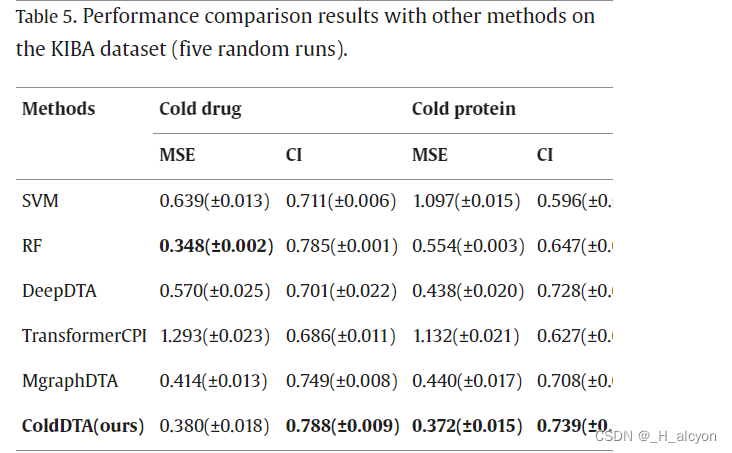

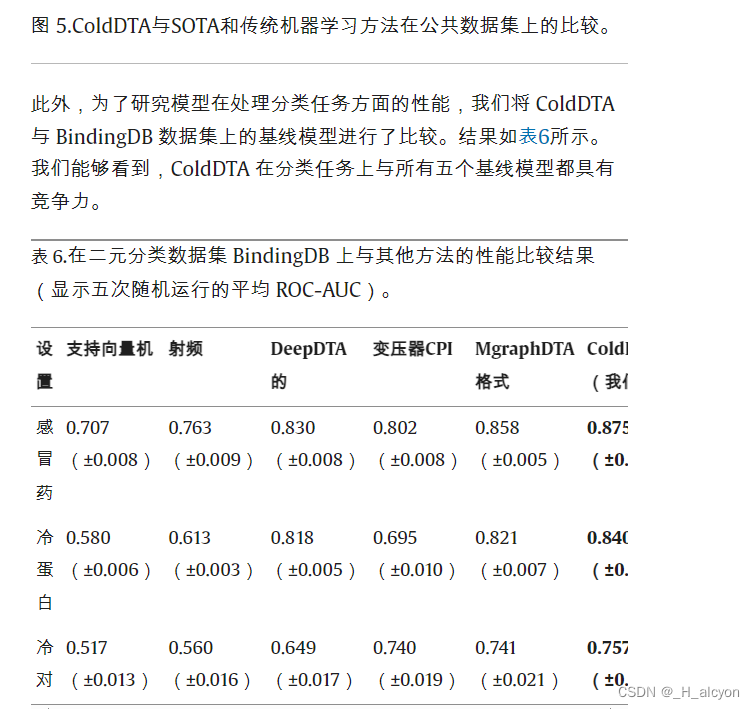
4.消融研究
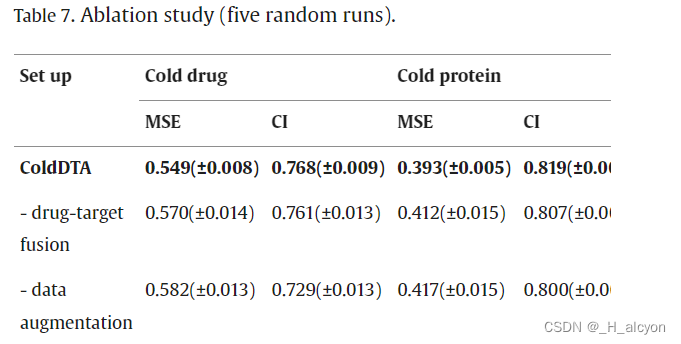
在Davis数据集上进行了消融研究,以验证所提出的数据增强策略和药物-靶点融合模块的有效性。我们使用了以下设置:
-
药物-靶点融合:我们用简单的串联操作取代了药物-靶点融合模块。
-
数据增强:我们进一步不使用数据增强,在这一点上,模型没有使用本文中提出的任何方法。
实验结果如表7所示,从中可以看出,基于注意力的特征融合和数据增强策略都有助于模型的预测性能。
此外,我们分别研究了数据增强对模型预测性能的影响,这部分实验都使用了药物-靶点融合模块。基准数据集中使用数据增强的最优参数如表8所示,由于超参数搜索空间范围有限,这些参数是通过网格搜索得到的,去除子图比率的搜索范围为[0.1,0.2,0.3,0.4],使用数据增强次数的搜索范围为[1, 2, 3, 4, 5].是否使用数据增强对预测性能的影响如图 6 所示,其中误差线的长度表示标准差。从图6可以看出,使用本研究提出的数据增强策略可以有效增强模型在两个公共数据集的感冒药和感冒配对任务中的预测性能。具体而言,在没有数据增强策略的情况下使用药物-靶点融合模块时,Davis感冒药、Davis冷药、KIBA感冒药、KIBA冷药的CI值分别为0.757±0.012、0.684±0.018、0.776±0.013、0.653±0.023。使用数据增强后,所有CI值均显著改善,相应的MSE值降低。但对于冷蛋白任务,我们的策略没有显著影响,在KIBA数据集中,它甚至有轻微的负面影响,CI从0.011变为0.739±0.011。本研究表明,数据增强方法对DTA回归任务是有效的,选择合理的方法可以有效提高模型的泛化能力。删除子图以形成药物和原始靶点之间结合亲和力不变的数据对似乎违背了常识,但实际上,多个数据增强以生成不同的子图允许训练集隐含包含完整的分子图谱信息,而深度学习模型能够更好地从剩余的子图中学习关键信息, 正如我们的实验所证明的那样。同时,人们也应该意识到,在少数情况下,使用完全随机的数据增强策略可能会产生非物理样本,类似于计算机视觉中的许多数据增强方法[42\u201243],我们将在未来的工作中尝试解决这个问题。
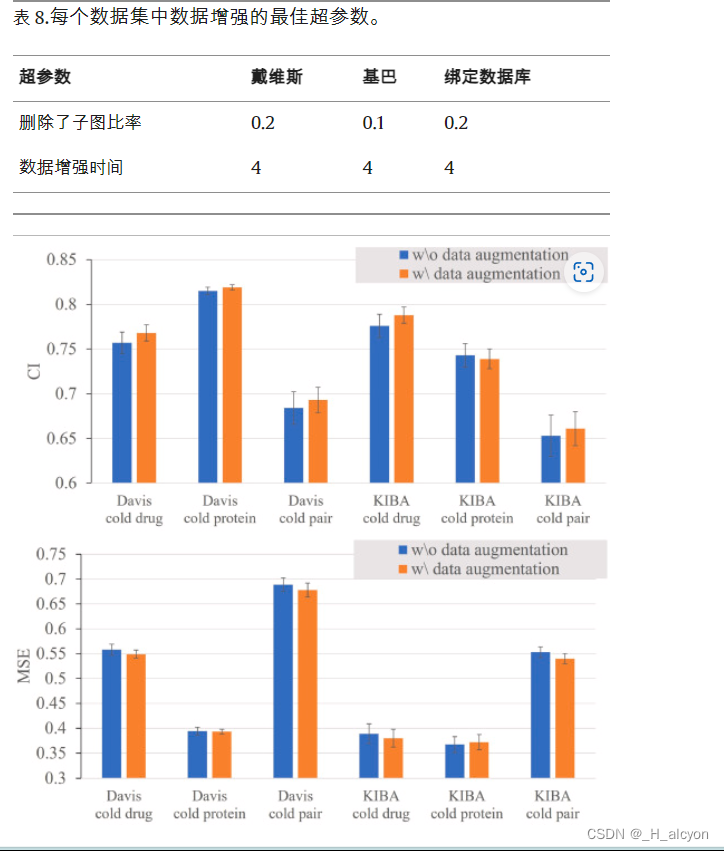
图 6.带或不带数据增强(五次随机运行)的模型性能。
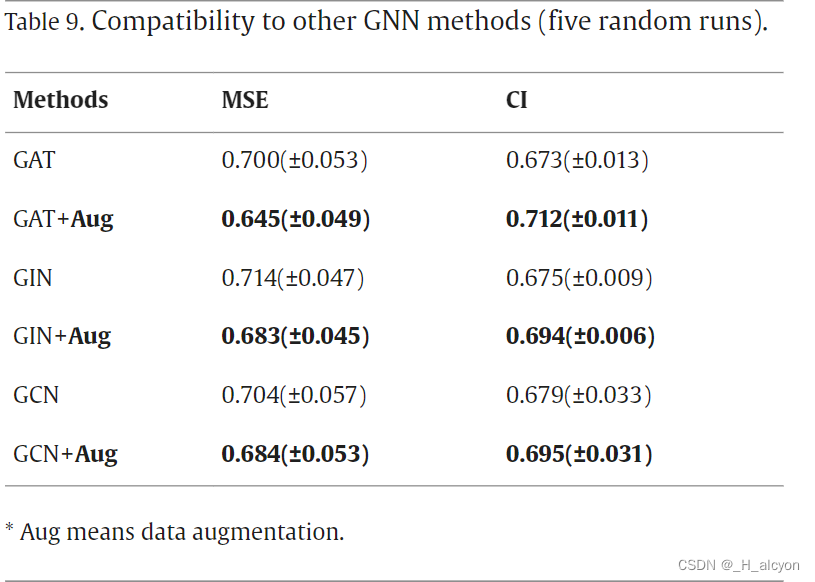
我们还通过实验证明了我们提出的数据增强策略的兼容性,其中我们选择图注意力网络 (GAT)、图同构网络 (GIN) 和图卷积网络 (GCN) 作为药物的特征编码器。这些实验是在Davis的感冒药分裂上进行的,因为我们的策略最适合感冒药,并且与KIBA数据集相比,训练时间更短。实验结果如表9所示,表明我们的数据增强方法与各种GNN网络兼容性较好。
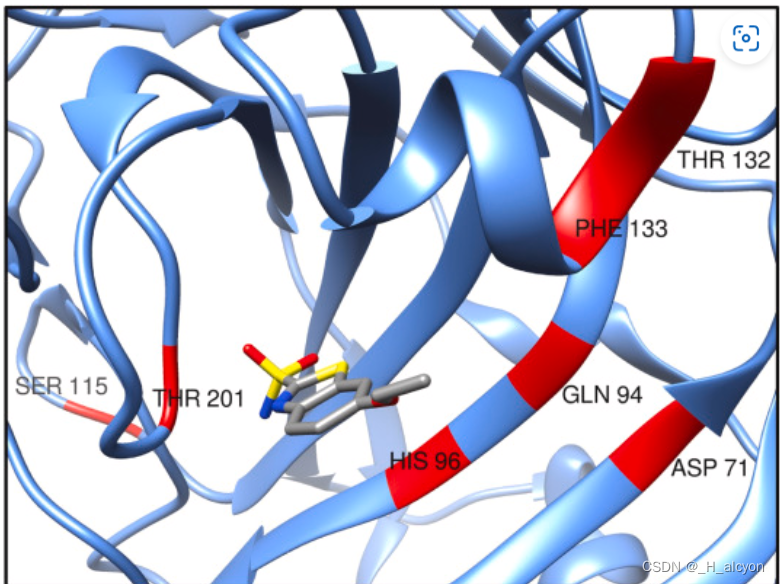
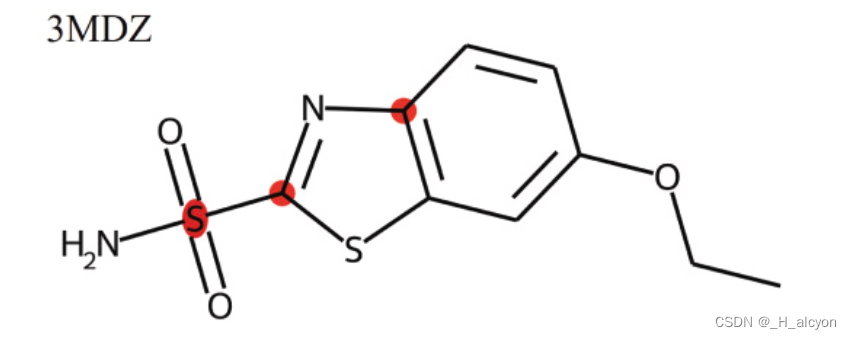
5.模型解释

总结
在这项研究中,我们提出了一种使用数据增强和基于注意力的特征融合来预测药物-靶标结合亲和力的端到端训练方法:ColdDTA。去除了训练阶段固定比例的药物分子图,与原目标形成新的数据对,这种数据增强可以有效提高模型的泛化能力。同时,该模型采用一种新的基于注意力的特征融合方法,使化合物与蛋白质信息更好地相互作用,从而提高了模型预测性能。实验结果表明,与SOTA基线和传统机器学习方法相比,ColdDTA在基准数据集上的性能显著提高。烧蚀实验也验证了所提方法的有效性。此外,我们还通过权重的可视化演示了模型的可解释性。