文献介绍

「文献题目」 SPACEL: deep learning-based characterization of spatial transcriptome architectures
「研究团队」 瞿昆(中国科学技术大学)
「发表时间」 2023-11-22
「发表期刊」 Nature Communications
「影响因子」 16.6
「DOI」 10.1038/s41467-023-43220-3
摘要
空间转录组学(ST)技术可以检测单个 cells/spots 中的 mRNA 表达,同时保留它们的二维(2D)空间坐标,使研究人员能够研究组织中转录组的空间分布;然而,联合分析多个 ST 切片并将它们对齐以构建组织的三维(3D)堆叠仍然是一个挑战。在这里,作者介绍了用于 ST 数据分析的空间结构特征化深度学习(SPACEL)。SPACEL 包括三个模块:Spoint 嵌入了一个带有概率模型的多层感知器,用于对单个 ST 切片中每个 spot 的细胞类型组成进行反卷积;Splane 采用了图卷积网络方法和对抗性学习算法来识别跨多个 ST 切片上转录组和空间上一致的空间域;Scube 自动转换连续切片的空间坐标系统,并将它们叠加在一起以构建组织的三维结构。使用来自各种组织的模拟和真实 ST 数据集和 ST 技术与 19 种最先进的方法进行比较,表明 SPACEL 在细胞类型反卷积、空间域识别和 3D 对齐方面优于其他方法,从而展示了 SPACEL 作为一种用于 ST 数据处理和分析的有价值的集成工具包。
研究结果
1. SPACEL 工作流程
SPACEL 的完整架构如 Fig. 1a 所示,包括模块 Spoint、Splane 和 Scube。作者开发 Spoint 模块的动机是需要对 seq-base ST 技术生成的 ST 切片进行细胞类型反卷积,这也是后续 SPACEL 分析所需要的。Spoint 嵌入了带有概率模型的多层感知器 (MLP),用于对每个 ST spot 的细胞类型组成进行反卷积(Fig. 1b, Methods)。Spoint 结合了模拟伪点、神经网络建模和表达谱的统计恢复,这使其能够提供更强大和更准确的框架来估计真实 ST 数据中的细胞类型比例。对于 image-based ST 数据(即单细胞分辨率),用户可以简单地使用单细胞分析工具(例如 Seurat 和 Scanpy)对细胞进行聚类,并使用标记基因来识别每个细胞类群的细胞类型。
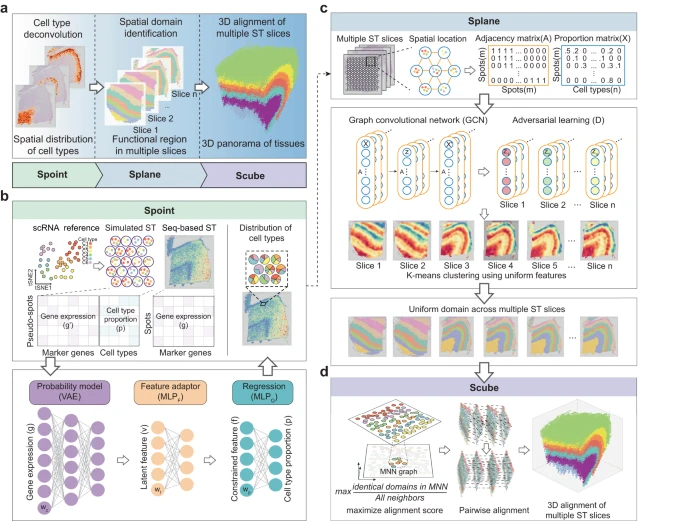
a.SPACEL 的三个模块:Spoint、Splane 和 Scube。
b.Spoint 使用 MLP 和概率模型对 spots 的细胞类型进行反卷积。MLP,多层感知器;VAE,变分自动编码器。
c.Splane 采用 GCN 模型和对抗性学习算法来识别跨多个切片的空间域。GCN,图卷积网络。
d.对于连续切片,Scube 采用相互最近邻 (MNN) 图和差分进化算法来变换切片并构建组织的堆叠 3D 对齐。
Splane 采用图卷积网络 (GCN) 方法和对抗性学习算法,通过联合分析多个 ST 切片来识别空间域(Fig. 1c,Methods)。首先,对于每个 ST 切片,Splane 根据 cells/spots 在空间中的距离计算一个邻接矩阵。然后,Splane 根据邻接矩阵和 cells/spots 的细胞类型组成构建 GCN 模型,并采用对抗性学习算法来学习所有分析的 ST 切片共享的潜在特征;作者将此称为联合分析方案。接下来,Splane 应用 K-means 聚类算法来聚合具有共享潜在特征的相似模式的 cells/spots,其中每个 cells/spots clusters 被称为一个空间域。所有现有的空间域识别工具通常都利用基因表达作为其分析的输入,并且大多数都遵循单一的分析方案。Splane 的独特之处在于利用细胞类型组成作为输入并采用联合分析方案。此外,Splane 还采用对抗性训练来明确解决和消除多个切片的批次效应。
对于包含连续切片的 ST 数据集,Scube 对齐每对相邻切片的 Splane 预测坐标系,然后构建组织的堆叠 3D 架构,这是完全自动化的,不需要任何手动对齐(Fig. 1d, Methods)。简而言之,Scube 根据 spots 的坐标信息在两个相邻切片的 cells/spots 之间构建相互最近邻(MNN)图,并构建它们之间的对齐目标函数。该函数作为对齐切片的基础,并将对齐任务视为优化问题。与 PASTE 或 STAligner 等现有工具不同,Scube 专门设计用于通过结合惩罚项来处理部分重叠的切片,该惩罚项负责计算相邻切片中重叠 spots 的比例。然后,Scube 采用差分进化优化算法来搜索每个切片的最佳平移向量和旋转角度,并通过相应地变换每个切片的坐标来对齐它们。最后,Scube 堆叠转换后的切片以构建组织的 3D 结构。
综上所述,Spoint、Splane 和 Scube 依次完成了 ST 数据的三项分析任务:细胞类型反卷积、空间域识别和 3D 架构构建。作者以创新的方式仔细设计了每个模块,以解决当前最先进方法的潜在局限性(Supplementary Fig. 1)。具体来说,Spoint 采用了统计模型、伪点模拟、深度学习技术,并消除了参考数据和 ST 数据之间的差异。相比之下,其他工具常常会错过其中一个或多个功能。Splane 的独特之处在于它结合了细胞类型组成作为输入(任何其他工具都没有使用这一功能)以及 GCN 模型中的对抗性训练(这是一种首先用于减轻 ST 数据中的批次效应的常见方法)。Scube 采用独特的全局优化策略进行 3D 对齐,使其在同类产品中脱颖而出。虽然每个模块都可以独立运行,但它们也经过优化,可以在统一的工作流程中无缝工作。这种适应性使 SPACEL 能够分析来自不同实验平台的数据,同时为 ST 数据解释提供全面且简化的解决方案。
为了确保 SPACEL 的鲁棒性,作者进行了广泛的实验来评估其在各种超参数设置下的性能。作者的结果表明,与其他最先进的方法相比,Spoint、Splane 和 Scube 对超参数变化表现出卓越的鲁棒性(Supplementary Fig. 2a–c),突显了 SPACEL 在不同实验中提供可靠且一致的结果方面的有效性设置。
2. Spoint 准确反卷积细胞类型组成
为了构建 Spoint 和其他反卷积方法的训练集,作者通过假设细胞数量和每个 spot 的细胞类型数量呈正态分布,使用 scRNA-seq 数据集模拟了 pseudo-spots。作者的模拟生成了与真实 ST 数据类似的 pseudo-spots(来自人脑组织的 MERFISH 数据;Supplementary Fig. 2d–k)。作者使用基准研究中的 32 个模拟数据集,将 Spoint 的性能与最先进的细胞类型反卷积方法(包括 Cell2lacation、SpatialDWLS、RCTD、STRIDE、Sterescope、Tangram、DestVI、Seurat、SPOTlight 和 DSTG)进行了比较(Fig. 2a, Supplementary Data 2)。作者采用皮尔逊相关系数(PCC)和结构相似性指数测量(SSIM)来评估预测细胞类型组成与真实细胞类型组成之间的相似性,并使用均方根偏差(RMSE)和 Jensen-Shannon 散度(JSD)来评估每个方法的误差。在 11 种反卷积方法中,Spoint 产生了最高的平均 PCC/SSIM 值(= 0.73/0.69)和最低的平均 RMSE/JSD 值(=0.05/0.41)(Fig. 2a)。此外,作者应用基准研究中定义的准确度得分(AS)来评估每种方法的性能:Spoint 的平均 AS(= 0.93)明显高于其他方法(AS = 0.24-0.82;Fig. 2b)。

a.基准研究中 32 个模拟数据集的反卷积方法的平均 PCC、SSIM、RMSE 和 JSD 值;PCC,皮尔逊相关系数; SSIM,结构相似性指数度量;RMSE,均方根误差;JSD,Jensen-Shannon 散度。
b.32 个模拟数据集的反卷积方法的准确度分数。
c.通过 DLPFC 数据集的反卷积方法预测的 excitatory layer 3/4 RORB+RPS3P6+ 神经元的空间分布。
d.DLPFC 数据集的反卷积方法的准确度分数。
为了评估 Spoint 在真实 ST 实验数据上的细胞类型反卷积方面的性能,作者使用了两个数据集作为输入:人类背外侧前额叶皮层 (DLPFC) 的 10X Visium 数据集,其中包含 12 个 ST 切片,以及从 Allen Brain Map 下载的人类大脑的 scRNA-seq 数据集。由于没有实验证据证明该数据集中每个 spot 的细胞类型组成,作者将原始研究注释的皮质层作为 ground truth(Fig. 2c, Supplementary Fig. 3a–c)。作为一个例子,作者发现 Spoint 预测 (AS = 0.85) 的兴奋性 L3/4 神经元的分布具有比其他实验的反卷积方法更高的 AS 值(AS = 0.29-0.64;Fig. 2c)。总的来说,对于所有 12 个 ST 切片中的所有 56 种细胞类型,Spoint 的平均 AS (= 0.60) 再次高于其他方法(平均 AS = 0.30-0.48;Fig. 2d)。此外,作者计算了各层之间一种细胞类型比例的任何差异的显着性(使用 Wilcoxon Rank Sum 检验),较低的 P 值表示反卷积精度提高。作者发现 Spoint 的平均 P-values ( = 0.01) 低于其他方法(平均 P-values = 0.05–0.64;Supplementary Fig. 3d)。
为了进一步评估 Spoint 在真实 ST 数据集中的性能,作者收集了三个单细胞分辨率 ST 数据集:Chen et al. 小鼠胚胎脑 (Stereo-seq),Chen et al. 小鼠大脑 (Stereo-seq) 和 Fang et al. 人脑(MERFISH with 4000 genes)来模拟具有已知细胞类型组成和空间背景的 spot 水平 ST 数据。作者还从相同的组织类型中获得了相应的 scRNA-seq 数据作为参考。使用这些数据,作者将大约 10 个细胞聚合到每个 pseudo spot 中,创建三个不同的 spot 水平 ST 数据集(Supplementary Fig. 4a)。例如,作者观察到,与数据集 1 中前脑谷氨酸神经母细胞的其他细胞类型反卷积方法相比,Spoint 实现了最高的 PCC(Supplementary Fig. 4b)。然后,作者使用四个评估指标评估了 Spoint 模块对三个数据集的所有细胞类型的反卷积性能。作者的研究结果表明,Spoint 始终优于现有方法,在所有三个数据集中实现了最高的准确度分数(Supplementary Fig. 4c, d)。此外,作者评估了预测细胞类型比例总和与真实值之间的相对误差,发现 SpatialDWLS(排名为 1、2 和 3)和 Spoint(排名为 4、3 和 1)记录了三个数据集的平均相对误差最低(Supplementary Fig. 4e)。这些结果有力地证明了 Spoint 模块相对于其他方法的优越性能,并且准确的反卷积结果将有助于后续使用 Splane 识别空间域。
3. Splane 识别多个切片的空间域
在使用 Spoint 对细胞类型进行反卷积后,作者应用 Splane 来识别上述 DLPFC 数据集 ST 切片的空间域(Supplementary Fig. 5)。作者使用原始研究的皮质层手动注释作为 ground truth,并使用三个指标来评估预测性能。具体来说,作者使用 Splane 识别的空间域与 ground truth 中相应的皮质层之间的 Jaccard 指数 (JI) 和调整兰德指数 (ARI) 来评估该方法的准确性,并计算预测和 ground truth 之间的移动距离 (SD) 来量化 Splane 的误差(see Methods)。较高的 JI/ARI 值或较低的 SD 值表明预测更接近 ground truth。
首先,作者将联合处理所有切片(joint analysis, by default)获得的结果与单独处理每个切片(single analysis)获得的结果进行比较。为了进行这种比较,作者构建了 Splane 的测试版本,即 Splane-single,它为每个切片单独构建了一个 GCN 模型。以 DLPFC 数据集中的切片 151508、151510、151670 和 151673 为例,联合分析得到的 JI 值高于单独分析得到的值,联合分析得到的 SD 值低于单独分析得到的值(Fig. 3a, b)。此外,在处理整个 12 个切片数据集时,联合分析的 average JI/median ARI 值为 0.61/0.61,高于单一分析的值(= 0.53/0.57;Fig. 3c, d);联合分析的平均 SD 值为 50μm,远低于单一分析的平均 SD 值(= 262 μm;Fig. 3e)。这一比较表明联合分析方案提高了 Splane 空间识别的准确性。
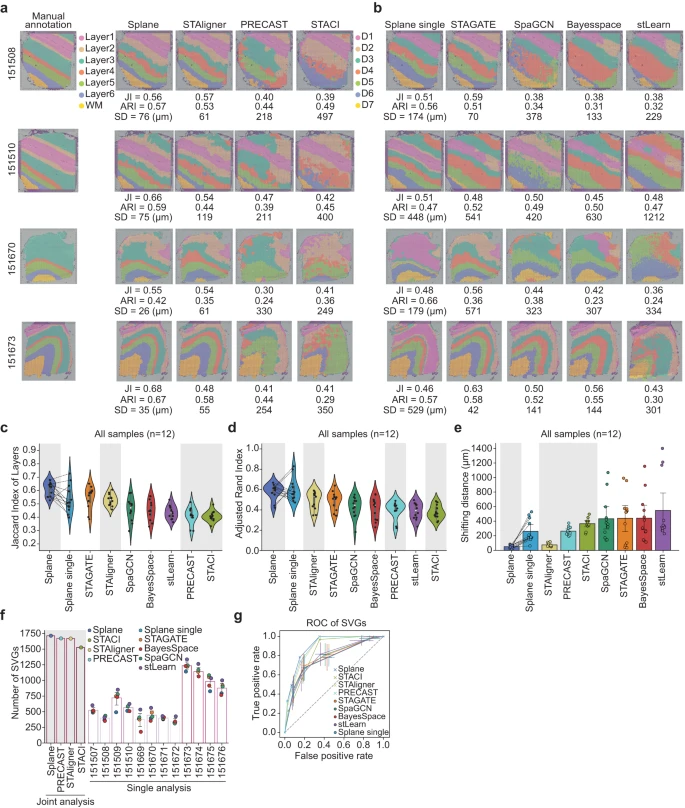
a.Splane、STAligner、PRECAST 和 STACI 对切片 151508、151510、151670 和 151673 识别的空间域的比较。Layer1~Layer6,皮质层 1 ~ 6;WM,白质;JI,Jaccard index;SD,shifting distance。
b.由 Splane-single、STAGATE、SpaGCN、BayerSpace 和 stLearn 为四个切片识别的空间域。
c-e.Jaccard 指数 (c)、调整后的 Rand 指数 (d) 以及皮质层与由 Splane、STAligner、PRECAST、STACI、Splane-single、STAGATE、SpaGCN、BayesSpace 和 stLearn 识别的相应空间域之间的移动距离 (e)。灰色背景代表多切片分析方法。
f.使用 fold-change > 0.5 且 P-value < 0.01(two-sided Wilcoxon rank-sum test)作为截止值时,通过空间域识别方法识别出的 SVGs 的比例。灰色背景代表用于多切片分析的方法。SVG,空间变化基因。
g.由 Splane、STAligner、PRECAST、STACI、Splane-single、STAGATE、SpaGCN、BayesSpace 和 stLearn 识别的 SVGs 的接受者操作特性 (ROC) 曲线。
然后,作者将 Splane 的性能与其他最先进的空间域识别方法进行了比较,包括设计用于分析多个 ST 切片的方法,如 STAligner、PRECAST 和 STACI,以及设计用于分析单个 ST 的方法,如 STAGATE、SpaGCN、BayesSpace 和 stLearn,使用相同的评估标准(Supplementary Data 2)。在分析 DLPFC 数据集的所有 12 个 ST 切片时,Splane 表现出最高的准确度(平均 JI/median ARI = 0.61/0.61)和最低的误差(平均 SD = 53 μm),而其他工具的 JI/ARI 和 SD 范围为 0.41–0.56/0.36–0.54 和 75–548 μm(Fig. 3a–e, Supplementary Fig. 6)。因此,Splane 优于空间域识别的可用替代方法。
基因的空间表达变异可以反映细胞的状态、通讯和动态,因此从 ST 切片中准确识别空间可变基因(SVGs)对于确定细胞在空间域中的功能和表型至关重要。因此,作者比较了这些方法来识别代表特定皮质层的 SVGs 的任务。作者采用原始研究注释的皮质层 1~6 和白质 (WM) 的 SVGs 作为 ground truth,并通过计算识别的 SVGs 与 ground truth 之间的重叠来评估每种方法的性能。使用 fold-change > 0.5 和 P-value < 0.01 作为 SVGs 的截止值,作者发现设计用于分析多个 ST 切片的工具比设计用于分析单个 ST 切片的工具识别出更多的SVGs。Splane、PRECAST、STAligner 和 STACI 在皮质层 1 ~ 6 和 WM 的 1917 个 SVGs 中识别出 1714、1671、1669 和 1527 个 SVGs,而其他检查方法在应用相同的截止值时,每个切片仅识别出 179 ~ 1336 个 SVGs(Fig. 3f)。同样,使用受试者操作特征(ROC)曲线作为 SVG 预测准确性的指标,作者发现 Splane、PRECAST 和 STAligner 产生的曲线下面积最高(AUC = 0.90、0.89 和 0.88),相比于所有检查的方法(AUCs < 0.83; Fig. 3g)。这些结果强调了 Splane 联合分析方案从多个 ST 切片中识别 SVGs 的能力。
为了评估 Spoint 模块在使用 Splane 模块促进准确空间域识别方面的重要性,作者对人类 DFPLC 数据集进行了两次比较分析。首先,作者使用三种不同的输入类型比较了 Splane 的性能:高度可变基因表达矩阵、高度可变基因表达矩阵的 PCA 降维、以及 Spoint 预测的细胞类型比例。结果表明,仅使用 Spoint 预测的细胞类型比例作为输入,产生的空间域识别结果与 ground truth 非常相似,与其他输入类型相比,所有 12 个切片中的 JI 值显着更高(Supplementary Fig. 7)。其次,作者使用其他方法的细胞类型反卷积结果作为输入来评估 Splane 的性能。结果表明,使用 Spoint 预测的细胞类型比例产生了最佳的空间域识别结果(Supplementary Fig. 8)。值得注意的是,只有使用从 Spoint 预测的细胞类型比例作为输入时,才可能识别 Layer4。
4. Splane 识别癌症切片的空间域
为了测试 Splane 在识别疾病切片空间域方面的性能,作者应用 Splane 联合分析了来自三个实验批次的 11 个乳腺癌 10X Visium 切片,其中包括参考文献中报告的 6 张切片,10X Genomics 发布的 4 张切片,以及参考文献中研究的 1 张切片。作者首先使用 Spoint 对每张切片进行细胞类型反卷积,并获得细胞类型身份(基于所选标记基因的表达)和每个 spot 的组成(Supplementary Fig. 9)。
随后,作者应用 Splane 并在 11 张切片中识别出 10 个空间域(Fig. 4a)。值得注意的是,与原始数据和没有对抗性学习的 Splane 相比,Splane 中不同实验切片之间的批次效应程度显着降低(Supplementary Fig. 10)。然后,作者探索了每个预测空间域的细胞类型组成,并相应地注释了这些 domains:domains D0-3、D4-6 和 D7-9 分别被定义为肿瘤(肿瘤细胞富集)、中间(混合细胞类型)和免疫(免疫细胞富集)domains(Supplementary Fig. 11a–d, Supplementary Data 3)。
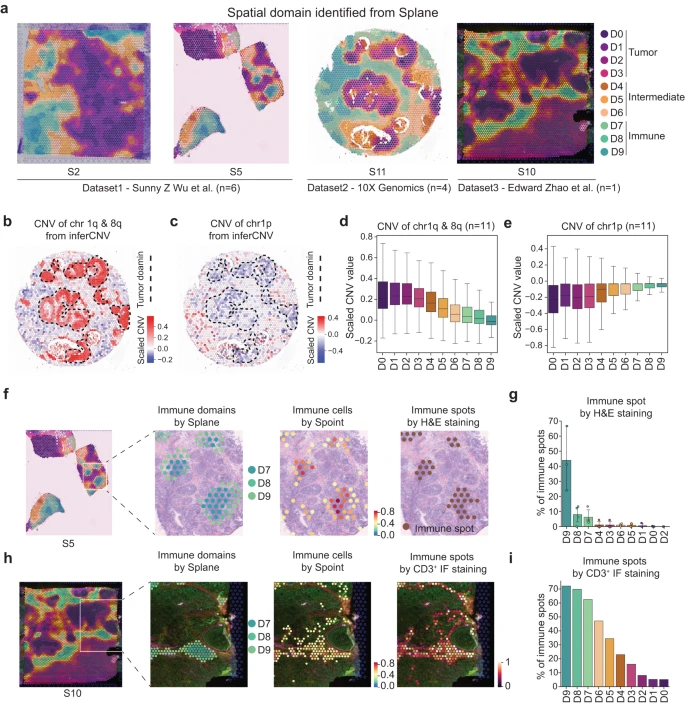
a.来自 Wu et al. 数据集的切片 S2 和 S5、Zhao et al. 数据集的 S10、以及 10X Genomics 发布的切片 S11 通过 Splane 识别的空间域。
b-c.切片 S11 中 ST spots 的染色体 1q&8q 拷贝数增加 (b) 和 1p 拷贝数损失 (c) 的空间分布,由 inferCNV 计算。虚线代表肿瘤域。
d-e.由 inferCNV 计算得到的每个空间域中染色体 1q & 8q (d) 和染色体 1p (e) 的 CNV。CNV,拷贝数变异。
f.从左到右:Splane 预测切片 S5 中的空间域、Splane 预测免疫域 D7/D8/D9 的分布、Spoint 预测免疫细胞的分布以及 H&E 染色标记免疫点的分布。
g.切片 S1、S2、S5 和 S6 的每个域中 H&E 染色标记免疫点的百分比。在最初的研究中,这四个切片进行了 H&E 染色。
h.从左到右:Splane 预测切片 S10 中的空间域,Splane 预测免疫域 D7、D8 和 D9 的分布,Spoint 预测免疫细胞的分布,以及 CD3+ 免疫荧光 (IF) 染色标记的免疫 spots 的分布。
i.切片 S10 的每个结构域中 CD3+ IF 染色标记免疫点的百分比。
然后,作者应用两种不同的方法来评估肿瘤和免疫域的 Splane 预测的准确性。由于乳腺癌患者的肿瘤细胞通常携带染色体拷贝数变异 (CNVs),包括染色体 1q 和 8q 增加和/或染色体 1p 丢失,作者的第一个方法是计算每个 ST spot 的 CNVs 并检查 CNVs 的富集情况在预测的肿瘤区域中。使用 inferCNV,作者根据 ST 数据的表达矩阵计算了所有 11 个 ST 切片中所有点的 CNVs。以切片 11 为例,Splane 预测的四个肿瘤结构域在 1q 和 8q 染色体上有拷贝数增加(推断的 CNV ≥ 0.2),以及在 1p 染色体上拷贝数丢失(推断的 CNV ≤ −0.2;Fig. 4b, c)。相反,在 Splane 预测的中间域或免疫域中检测到较少或没有拷贝数增加或丢失(推断的 CNV = −0.2 ~ 0.2)。将所有 11 个切片的平均 CNV 值考虑在内,作者还观察到肿瘤区域中染色体 1q 和 8q 增加(平均 CNV > 0.2)和染色体 1p 损失(平均 CNV < −0.2),但中间区域较少,免疫区域没有(Fig. 4d, e)。
作者的第二种方法是计算实验注释的免疫 spots 的百分比并检查空间域中免疫 spots 的富集情况。作者从参考文献中获取了切片 S1、S2、S5 和 S6 中 H&E 染色标记的淋巴细胞富集 spots 和参考文献中 S10 切片中 CD3+ 免疫荧光 (IF) 标记的 T 细胞富集 spots 作为免疫 spots 的 ground truth。然后作者计算了每个 Splane 识别的域中免疫 spots 的百分比。作者发现切片 S1、S2、S5 和 S6 中免疫域 D9、D8 和 D7 中 H&E 染色标记的免疫 spots 的百分比(分别为 44%、8% 和 6%)高于中间切片和肿瘤中的免疫 spots 域 ( < 1%)(Fig. 4f, g)。同样,在切片 S10 中,作者在免疫域 D9、D8 和 D7 中发现了更多的 CD3+ IF 标记的免疫 spots(分别为 72%、70% 和 63%),而在中间域和肿瘤域中则更多( < 47%)(Fig. 4h, i)。这些结果支持了 Splane 对肿瘤和免疫域的预测。
在肿瘤系统的背景下,准确识别肿瘤边界对于免疫肿瘤学研究至关重要。作者比较了 Splane 和其他多切片分析算法(包括 STAligner、PRECAST 和 STACI)识别肿瘤边界的能力。在人类乳腺癌 10X Visium 数据中,只有 Splane 准确识别了具有清晰边界的肿瘤区域(Supplementary Fig. 12a)。Splane 预测的肿瘤区域与特定染色体中 CNV 的增加或减少一致,并且与 H&E 图像中明显的肿瘤组织区域对齐良好。此外,作者观察到肿瘤细胞比例与 CNV 评分之间存在显着相关性,特别是在 Splane 识别的空间域中(Supplementary Fig. 12b)。这些结果凸显了 Splane 识别肿瘤系统中多个切片的一致肿瘤区域和边界的能力。
5. Scube 从连续的 2D 切片构建组织的 3D 结构
目前大多数实验 ST 技术测量的是转录本在 2D 空间中的分布,但组织显然是 3D 的,并且细胞的功能是在空间中分布的。因此,作者开发了 SPACEL 的 Scube 模块来构建和研究给定组织的 3D 架构。作者首先使用了参考文献中的小鼠大脑 3D STARmap 数据集(最初为 1400 μm × 1700 μm × 100 μm)并将其分成十个厚度为 10 μm 的切片作为 ground truth(Fig. 5a)。作者随机旋转、翻转和裁剪每个切片,以模拟包含连续 2D 切片的扰动 ST 数据集(Fig. 5b, see Methods)。作者将随机裁剪的切片的百分比定义为该切片的裁剪比率。在每个裁剪比例级别,作者使用 Splane 来识别十个切片的空间域,并应用 Scube 通过基于 Splane 识别的空间域转换它们的坐标系来对齐这些切片。
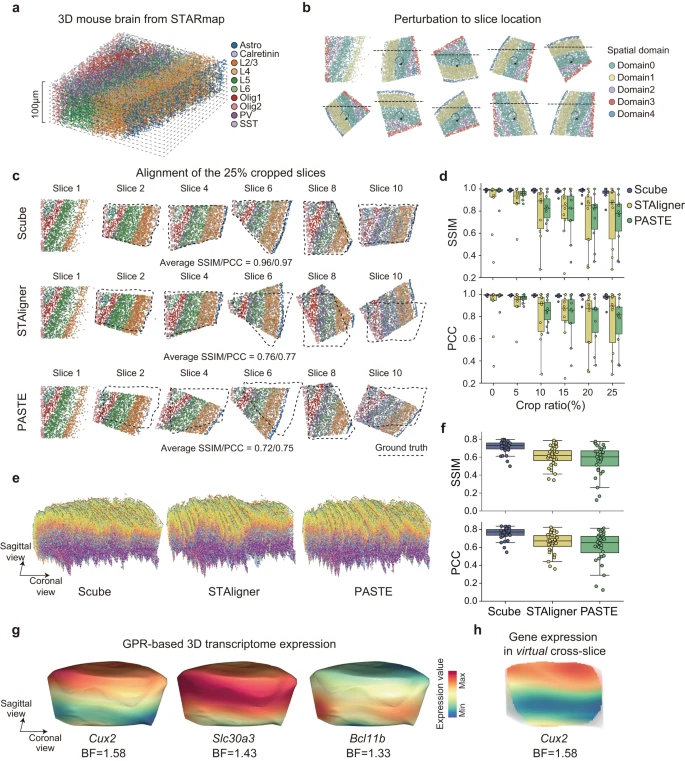
a.小鼠大脑的 3D STARmap 数据集。
b.通过将 3D STARmap 数据集划分为 10 个 2D 切片并随机旋转/翻转/裁剪每个切片,用连续 2D 切片模拟 ST 数据集。
c.裁剪比例=0.25 的模拟数据集的对齐,由 Scube、STAligner 和 PASTE 生成。虚线代表 ground truths 的位置。SSIM,结构相似性指数度量;PCC,皮尔逊相关系数。
d.裁剪比率从 0 到 0.25 的模拟数据集的 Scube、STAligner 和 PASTE 结果的 SSIM/PCC 值。
e.由 Scube(左)、STAligner(中)和 PASTE(右)从小鼠初级运动皮层的 33 个 MERFISH 切片构建的堆叠 3D 对齐。
f.MERFISH 数据集的 Scube、STAligner 和 PASTE 对齐结果的 SSIM/PCC 值。
g.在 Scube 中应用 GPR 模型后,基因 Cux2、Slc30a3 和 Bcl11b 在小鼠大脑 3D 结构表面的连续表达分布。 BF,贝叶斯因子。
h.Cux2 在小鼠大脑 3D 结构的冠状面上的连续表达分布。BF,贝叶斯因子。
作者采用 SSIM 和 PCC 作为指标来评估转换后的坐标系与每个切片的 ground truth 之间的相似性。SSIM 和 PCC 值越高表示对齐效果越好。回想一下,STAligner 和 PASTE 是两种最先进的方法,用于从连续 ST 切片构建组织 3D 对齐,作者将 Scube 与 STAligner 和 PASTE 的性能进行比较,以在裁剪比率从 0 到 0.25 的情况下对齐模拟数据集。由于 STAligner 和 PASTE 的对齐结果可能会受到其超参数的影响,因此在与 Scube 进行比较之前,作者首先进行了参数优化搜索,以实现 STAligner 和 PASTE 的最佳对齐结果(Supplementary Fig. 13)。当裁剪比等于 0.25 时,Scube 转换切片的平均 SSIM/PCC 值为 0.96/0.97,而 STAligner 转换切片的值为 0.76/0.77,PASTE 转换切片的值为 0.72/0.75(Fig. 5c) 。在裁剪比例范围 = 0.10、0.15、0.20 和 0.25 的情况下,Scube 转换切片的平均 SSIM 值分别为 0.98、0.98、0.97 和 0.96,高于 STAligner 的结果( = 0.79、0.79、0.72 和 0.76)或 PASTE(= 0.82、0.77、0.74 和 0.72;Fig. 5d)。同样,Scube 转换切片的平均 PCC 值为 0.99、0.98、0.98 和 0.97,也高于 STAligner( = 0.80、0.81、0.73 和 0.77)或 PASTE( = 0.85、0.79、0.76 和 0.75;Fig. 5d)的结果。表明 Scube 中的模拟数据集有更好的对齐方式。
为了进一步比较 Scube、STAligner 和 PASTE 在真实 ST 数据集上构建 3D 架构时的性能,作者检查了小鼠初级运动皮层 (MOp) 轮廓,其中包含来自 MERFISH 实验的 33 个连续 2D 切片。作者首先应用 Splane,从 33 个连续的 2D 切片中识别出 7 个空间域,它在空间域识别中表现出最高的准确度(平均 JI/median ARI = 0.44/0.43),相比于 STACI(平均 JI/median ARI = 0.43/0.38)、STAligner(平均 JI/median ARI = 0.33/0.25)和 PRECAST(平均 JI/median ARI = 0.23/0.16;Supplementary Fig. 14)。然后,作者使用 Scube 通过转换每对相邻切片的坐标系来对齐这些切片(Supplementary Fig. 15a, b)。接下来,通过将转换后的 2D 切片堆叠在一起,作者在 Scube 中构建了 MOp 的 3D 架构,从而能够对空间域和细胞类型进行 3D 说明(Fig. 5e)。
同时,作者应用 STAligner 和 PASTE 从同一数据集执行对齐和 3D 构建(Supplementary Fig. 15)。由于在这种情况下不知道 ground truth,作者计算了每两个相邻变换切片之间的 SSIM/PCC 值来评估两种方法的对齐性能,发现 Scube 变换切片的平均 SSIM/PCC 值为 0.71/0.76,显着高于 STAligner (SSIM/PCC = 0.61/0.65) 或 PASTE (SSIM/PCC = 0.57/0.61;Fig. 5f) 的结果。值得注意的是,当使用 STAligner 或 PASTE 堆叠 2D 转换切片时,作者获得了 MOp 的扭曲 3D 架构(Fig. 5e)。
SPACEL 还在 Scube 中集成了高斯过程回归 (GPR) 模型,该模型可以预测 3D 架构中任何位置的基因表达水平,从而能够连续说明沿空间任何方向的转录本分布(Fig. 5g, Supplementary Fig. 16a, b)。通过量化 GPR 模型的贝叶斯因子(BF,参见方法),SPACEL 能够识别在组织 3D 空间内任何方向上显着变化的基因(Fig. 5h)。因此,SPACEL 允许用户从任何方向探索转录本分布的动态,从而揭示复杂组织或器官的空间架构的真实 3D 结构。
详细 3D 结构的准确重建对于揭示空间组学数据中的生物现象至关重要。在小鼠胚胎 Stereo-seq 数据中,与 STAligner 和 PASTE 相比,Scube 在准确重建和描绘大脑、肝脏和爪子等结构方面取得了更好的性能(Supplementary Fig. 17a, b)。此外,与 STAligner (0.57/0.61) 和 PASTE (0.24/0.25) 相比,Scube (SSIM/PCC = 0.62/0.66) 在切片之间的组织区域分布方面表现出更高的一致性(Supplementary Fig. 17c)。综上所述,SPACEL 的 Scube 模块在模拟(STARmap)和真实(MERFISH 和 Stereo-seq)数据集的对齐和 3D 架构构建方面均优于 STAligner 和 PASTE。
6. SPACEL 作为 ST 数据处理和分析的集成工具包
为了突出 SPACEL 的集成性质,作者应用完全集成的工作流程来分析小鼠全脑 ST 数据。该数据集由空间转录组学技术生成的 75 个连续切片组成,点分辨率为 100μm,覆盖了小鼠大脑的大部分区域。使用原始大脑区域注释作为参考 ground truth,作者评估了每个模块的性能。在 Spoint 中,主要区域特异性细胞类型的预测分布与相应的大脑区域非常匹配,例如兴奋性神经元以及主要预测在海马区域的海马 CA1 和 CA3 细胞(Supplementary Fig. 18a, b)。在 Splane 中,作者将其性能与 STAligner、PRECAST 和 STACI 进行了比较。在 75 个切片中,Splane 识别的空间域的平均 JI/median ARI 值 (0.42/0.56) 高于 STAligner (0.36/0.40)、PRECAST (0.35/0.38) 和 STACI (0.31/0.40) 识别的空间域(Supplementary Fig. 18c, d)。在 Scube 中,Scube (SSIM/PCC = 0.83/0.85) 实现的 3D 组织重建表现出较高的一致性和准确性,超过了 PASTE (0.82/0.84) 和 STAligner (0.79/0.81) 获得的结果(Supplementary Fig. 18e, f)。这些结果清楚地表明,SPACEL 可作为分析多个 ST 切片的有效集成工具包。
讨论
在本研究中,作者介绍了一个基于深度学习的工具包 SPACEL,包含三个模块:Spoint 用于细胞类型反卷积、Splane 用于跨多个 ST 切片识别空间域、Scube 用于从连续 ST 切片构建 3D 架构。通过分析使用五种不同 ST 技术获取的 32 个模拟 ST 数据集和 11 个真实 ST 数据集,作者证明了 SPACEL 模块在每项任务上都优于最先进的方法。虽然每个模块都可以单独使用,但它们在 SPACEL 平台内的协同相互作用提供了一个全面且简化的解决方案,可实现 ST 数据解释的最佳结果,特别是准确的 3D 组织对齐、精确的空间域识别和有效的批次效应消除。
与其他最先进的方法不同,SPACEL 的 Splane 模块将细胞类型组合作为输入和 GCN 模型中的对抗训练相结合。由于来自不同 ST 实验的基因、细胞和细胞类群的空间分布可能极其异质,因此使用细胞类型组成作为输入并向 GCN 模型引入对抗性训练可显着最小化批次效应,从而产生更稳健、更高效的空间域识别方法。Splane 所采用的联合分析方案对于识别共同特征也特别强大,因此可以准确预测跨多个切片的 SVGs。
Splane 中识别的空间域也为 Scube 实现连续切片的精确对齐以及精确的 3D 架构构建奠定了基础。相比之下,PASTE 算法使用原始表达矩阵和 ST 数据的空间坐标作为比对输入,并使用超参数 α 来调整比对点之间转录差异和空间距离的相对贡献。这种设计使得 PASTE 的对齐结果对具有重大缺陷的数据集中的超参数敏感(例如,裁剪比率 ≥ 0.25 的切片)。另一方面,STAligner 基于原始表达矩阵构建 MNN 以选择标志点/细胞,并依赖于跨切片共享的用户定义的标志域进行比对。然而,这种方法没有利用整个切片每个点的完整信息,这限制了其平衡全局结构相似性和对齐精度的能力,特别是在部分重叠切片的情况下。相比之下,Scube 采用全局优化策略进行 3D 对齐,考虑到相邻切片中所有点之间的对应关系。这种创新方法使 Scube 能够实现更精确的对齐,在空间对齐过程中保持整体结构的完整性。
为了增强 3D 空间转录组学 GPR 模型的可访问性,作者通过集成原始 MATLAB 代码和第三方 GUI 软件开发了用户友好的 Python 代码。这种集成使研究人员能够轻松访问和利用该模型,促进该领域的进步并促进科学界更广泛地采用该技术。有机会提高 SPACEL 的性能并扩展其应用。例如,Splane 使用细胞类型组成信息作为模型的输入;这意味着,要处理 seq-based 的 ST 数据集(例如 10X Visium),必须采用细胞类型反卷积算法(例如 Spoint)来获取细胞类型组成信息,然后才能应用 Splane。此外,当前版本的 Scube 不支持非线性对齐以及 STAligner 和 PASTE。尽管有多种工具可以通过手动设置锚点来非线性对齐图像数据,但据我们所知,目前还没有方法可以自动进行 ST 数据分析。作者承认这一限制,并计划在 Scube 的未来版本中解决此问题。SPACEL 的另一个问题是,当向数据集中添加新切片时,整个深度学习模型需要从头开始重新训练,这可能会影响 SPACEL 分析大规模数据集的计算效率。作者预计,已用于生物信息学任务(例如大规模单细胞数据的细胞类型分类)的迁移学习模型和算法(例如 ImageNet 和 BERT)可以帮助克服这一限制。
注:本文为个人学习笔记,仅供大家参考学习,不得用于任何商业目的。如有侵权,请联系作者删除。

本文由 mdnice 多平台发布